











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La hemofilia adquirida de tipo A (HAA) es una enfermedad hemorrágica poco común determinada por un trastorno de coagulación resultante de la generación de anticuerpos contra el factor VIII de la coagulación (FVIII).1 Estos autoanticuerpos o inhibidores neutralizan parcial o totalmente la funcionalidad del FVIII, o bien provocan su eliminación.2 Estos autoanticuerpos son heterogéneos, del tipo inmunoglobulina G (IgG); al parecer, se producen debido al polimorfismo del antígeno 4 de los linfocitos T citotóxicos, que provoca una alteración en el funcionamiento de la tolerancia periférica.3
La HAA tiene una incidencia de alrededor de 1.5 millones de personas cada año.4 Habitualmente, los episodios hemorrágicos ocurren de manera espontánea en 77.4% de los casos, y en 12.64% se relacionan con traumatismos, procedimientos quirúrgicos o incidentes durante el periodo periparto o postparto. Su presentación es más frecuente en las edades tardías de la vida y predomina en las mujeres. La presentación clínica es variable y puede manifestarse como púrpura (53.2%), sangrado severo de los músculos o retroperitoneo (50.2%), sangrado de las mucosas (31.6%), hemartrosis (4.9%) o sangrado intracerebral (1.1%).5
En general, los episodios de hemorragia aguda son graves en 70.3% de los casos; por lo tanto, el control puntual de la hemorragia es vital.6 Esta condición clínica ejemplifica un desafío médico exigente, ya que los individuos con HAA tienen eventos hemorrágicos más severos que los pacientes con hemofilia congénita con títulos similares del FVIII.7 Su diagnóstico se complica debido a que aquéllos con HAA no tienen antecedentes familiares de trastornos de coagulación; asimismo, no presentan un cuadro clínico similar al de la hemofilia hereditaria. Por tales motivos, se retrasa el diagnóstico, lo que incide en la elección del tratamiento específico.8 Esta condición clínica debe ser sospechada ante la presencia de hemorragias francas con tiempo prolongado de tromboplastina parcial activada (TTPa) y sin antecedentes familiares previos de trastornos de coagulación. Esta enfermedad debe ser siempre considerada ante una hemorragia inexplicable, debido a que su tasa de mortalidad es alta: oscila entre 7.9 y 22%.
Caso clínico
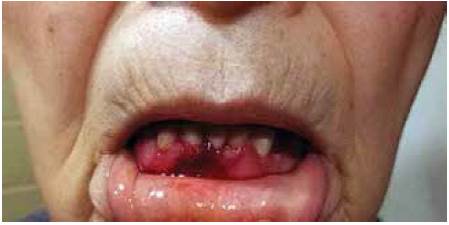
Paciente de 80 años de edad que acudió a consulta debido a la presencia de ampollas sangrantes en la cavidad oral, las cuales habían aparecido una semana antes de su ingreso. Tenía antecedente de diabetes tipo 2, diagnosticada desde hacía diez años y controlada con hipoglucemiantes orales y dieta. No tenía antecedentes familiares de trastornos de la coagulación. El examen físico mostró presencia de una ampolla sangrante en la cavidad oral (Figura 1A), ampollas sangrantes localizadas en la espalda y pecho (Figura 1B) y sangrado persistente en la encía inferior (Figura 2).
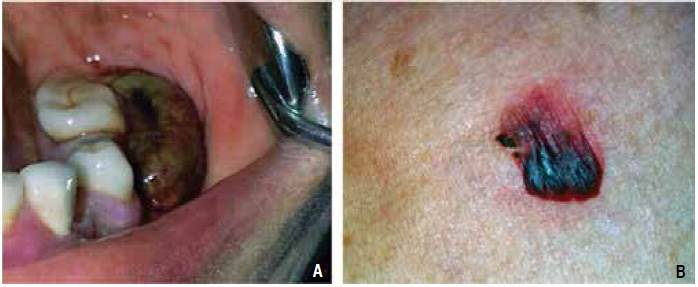
Figura 1 A. Ampolla con contenido hemático localizada en la región inferior posterior izquierda, ocupando el espacio vestibular. B. Ampolla hemática situada en la espalda.
Las pruebas de laboratorio informaron un TTPa de 66.6 segundos (24.3-35 segundos), no corrigió en la prueba de mezcla, con tiempo de protrombina (TP) normal. La FVIII:C fue de 2.57% (50-150%), los títulos de los inhibidores de la FVIII:C fueron de 89.6 BU/mL (< 0.5 BU/mL) y los niveles séricos de los factores de coagulación IX, X, XI, XII fueron normales. Por los resultados mencionados más la edad de la mujer y la ausencia de antecedentes familiares de hemorragia, se estableció el diagnóstico de HAA.
La paciente recibió tratamiento con el factor VII recombinante activado (rFVII), a razón de 90 μg/kg intravenosos (sólo un bolo) y prednisona (1 mg/kg/día) durante ocho semanas, asociada con ciclofosfamida (1 mg/kg/día) durante seis semanas. Los fármacos se interrumpieron cuando se detectó la normalización del TTPa. Durante un año de seguimiento, no presentó más sangrados.
Discusión
La HAA es una enfermedad poco común producida por autoanticuerpos IgG1 o IgG4 dirigidos contra epítopos dentro de los dominios A2, A3 y C2 del FVIII. La etiología de la HAA permanece incierta, así como la incidencia baja en relación con casos clínicos caracterizados por la presencia de sangrado en la cavidad oral.9 En general, este trastorno se asocia con condiciones autoinmunes, trastornos dermatológicos, neoplasias malignas hematológicas, infecciones por hepatitis B y C, y diabetes mellitus (Cuadro 1), como en el caso presentado.10
Cuadro 1 Enfermedades asociadas con la hemofilia adquirida tipo A.
| Enfermedades alérgicas | Asma |
| Medicamentosas | |
| Enfermedades autoinmunes | Artritis reumatoide |
| Colitis ulcerativa | |
| Esclerosis múltiple | |
| Lupus eritematoso sistémico | |
| Miastenia gravis | |
| Síndrome de Sjögren | |
| Enfermedades dermatológicas | Pénfigo |
| Psoriasis | |
| Enfermedades hematológicas | Leucemia linfocítica crónica |
| Macroglobulinemia de Waldenström | |
| Mielofibrosis | |
| Síndrome mielodisplásico | |
| Enfermedades oncológicas o tumores | Cabeza y cuello |
| Colón | |
| Linfoma no Hodgkin | |
| Mama | |
| Próstata | |
| Riñón |
Esta condición clínica debe asumirse cuando un paciente presenta una diátesis hemorrágica no reconocida, especialmente en adultos mayores o mujeres en el postparto. El diagnóstico se establece mediante estudios de laboratorio que incluyen una prolongación del TTPa, niveles reducidos de FVIII y la presencia de inhibidores de FVIII.11,12
El tratamiento de la HAA consiste en el control de la hemorragia usando rFVIIa o el complejo de protrombina activado pd-aPCC y terapia inmunosupresora.13,14 Para el control de los inhibidores se puede utilizar fármacos inmunosupresores como corticosteroides, ciclosporina, ciclofosfamida o azatioprina.15 La paciente respondió bien al tratamiento combinado de prednisona y ciclofosfamida asociadas con rFVIIa.
Conclusiones
La identificación inicial expedita y la aplicación del tratamiento son fundamentales para disminuir la tasa de mortalidad asociada con la HAA. Sin embargo, el diagnóstico de HAA es a menudo complicado debido a la ausencia de antecedentes familiares de sangrado y a la etiopatogenia distinta de este trastorno. Empero, un paciente podría ser identificado por un historial de episodios hemorrágicos inexplicables asociados con un TTPa prolongado. El rFVIIa o pd-aPCC debe usarse como terapia de primera línea para el tratamiento de esta condición clínica, asociado con terapia inmunosupresora para eliminar los inhibidores de FVIII.

