











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkANTECEDENTES
En el estudio de la pareja infértil, el del varón se inicia con el análisis seminal que permita identificar la azoospermia, que se encuentra, incluso, en 1% de la población general masculina.1 Como parte del estudio de estos pacientes debe realizarse una exploración física cuidadosa que incluya el habitus exterior, la testicular y las estructuras adyacentes. Cuando hay volúmenes seminales anormales, con azoospermia u oligoazoospermia, sin otras alteraciones anatómicas, hay que sospechar un origen no obstructivo, que invariablemente requirá perfil hormonal y estudios genéticos (microdeleciones y cariotipo). 2,3
El síndrome del varón 46 XX o síndrome De La Chapelle es una causa infrecuente de azoospermia. Se describió por primera vez en 1964 y su incidencia es de 1 caso por cada 20,000 recién nacidos vivos varones.2-9 Se trata de un trastorno en la diferenciación sexual en donde el fenotipo, las gónadas y el sexo cromosómico son discordantes.2 La mayoría de los casos se identifica en la consulta de infertilidad, en adolescentes y en adultos.5,10 El síndrome es secundario a la existencia del gen SRY, normalmente localizado en el brazo corto del cromosoma Y, que se transferirá hacia el cromosoma X. El gen SRY se encuentra en 80% de los casos de varón 46 XX, 4,6,9 por lo que cursan con pubertad normal, con caracteres sexuales secundarios, desarrollo de genitales internos y externos masculinos.10 Al parecer, el gen SRY es el encargado de codificar al factor determinante de la diferenciación testicular, cuya existencia y expresión son necesarias para inactivar las señales de diferenciación sexual femenina y activar la diferenciación sexual masculina.3,4,11 Este gen se localiza en el cromosoma Yp11.3,12 y quizá se transfiere al cromosoma X durante la meiosis paterna.4
Por lo que se refiere al funcionamiento reproductivo, estos pacientes tienen azoospermia debida a la espermatogénesis, mediada por células de Sertoli, y por ausencia de los factores de azoospermia localizados en distintas porciones del cromosoma Y, indispensables para la espermatogénesis normal.3,6,10 Las células de Leydig, aunque hipertrofiadas, tienen producción normal o disminuida de andrógenos. Por eso los pacientes pueden tener un desarrollo normal de caracteres sexuales secundarios masculinos.6,13
CASO CLÍNICO
Paciente de 37 años, con deseo de fertilidad, 1.62 m de estatura, peso de 55 kg, tensión arterial de 110-70 mmHg, frecuencia cardiaca de 80 x minuto y respiratoria de 20 x minuto, alopecia frontoparietal, genitales externos con pene normal, hipotrofia testicular bilateral. El testículo derecho de 1.5 cc y el izquierdo de 2 cc, Tanner III; el resto de la exploración física sin alteraciones y sin paternidad comprobada.
A la pareja se le practicaron estudios de laboratorio y gabinete. A la paciente se le solicitó progesterona, prolactina y hormona estimulante de tiroides (TSH) tomados en fase lútea media que indicaron ovulación, prolactinemia normal y eutiroidismo. La histerosalpingografía reportó que la cavidad uterina se encontraba normal y las salpinges permeables. Al varón se le solicitó un análisis seminal que reportó azoospermia; se repitió a las 2 semanas y nuevamente se encontró azoospermia, con un espermocultivo en búsqueda de Chlamydia y Mycoplasma que se reportó negativo. El semen se centrifugó para confirmar el diagnóstico de azoospermia. Los siguientes estudios se enfocaron en conocer la causa de la azoospermia y poder iniciar un tratamiento adecuado. El ultrasonido testicular reportó ambos testículos disminuidos de tamaño con contornos regulares, derecho 1.4 x 0.7 x 1.6 cm con volumen de 0.9 cc (Figura 1), izquierdo 1.6 x 1.5 x 0.8 cm con volumen de 1.1 cc (Figura 2). Epidídimos hipoecoicos con un quiste en la cabeza, en el lado derecho de 1.7 mm. Ambos cordones espermáticas sin flujo Doppler y disminución en su ecogenicidad.
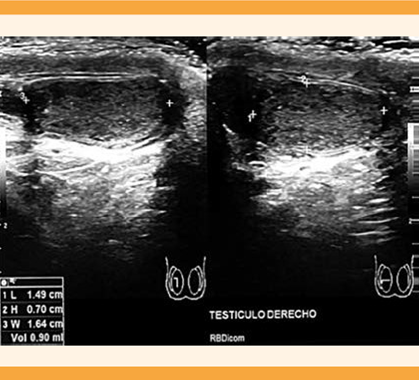
Figura 1 Imagen de ultrasonido testicular derecho, se observan los cortes longitudinal y transversal y el volumen testicular con hipotrofia.
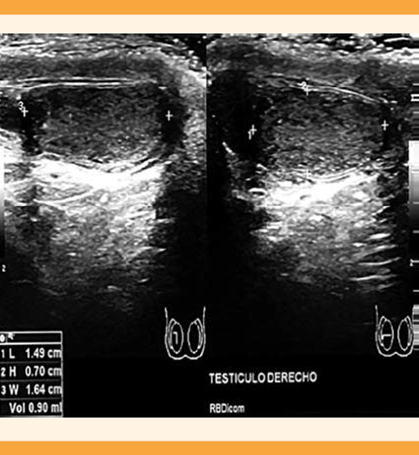
Figura 2 Imagen de ultrasonido testicular izquierdo en el que se observan cortes longitudinal y transversal, con diagnóstico de hipotrofia testicular.
En los estudios hormonales se reportó: hormona folículo estimulante (FSH): 58.13 mUI/mL (1.4-18.1 mUI/mL), hormona luteinizante (LH): 24.15 mUI/mL (1.5-9.3 mUI/mL), estradiol (E2): 21 pcg/mL (25-50 pc/mL), testosterona total (TT): 2.17 ng/mL (2.41-8.27 ng/mL), que integraron el diagnóstico de hipogonadismo hipergonadotrópico secundario a la insuficiencia testicular primaria asociada con diagnóstico de azoospermia no obstructiva. Para completar el protocolo se le realizó el estudio genético que reportó un cariotipo convencional, con bandas GTG, en linfocitos de sangre periférica. Se analizaron 40 metafases y se reportó un cariotipo femenino normal 46 XX. Se decidió repetir en ese momento el estudio, con igual reporte. Figura 3
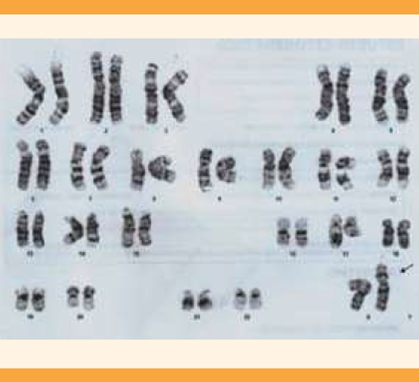
Figura 3 Cariotipo con bandas GTG en linfocitos de sangre periférica con reporte de cariotipo femenino 46 XX.
El estudio de hibridación in situ (FISH), en sangre periférica, corroboró la existencia del gen SRY y la traslocación del brazo corto del cromosoma X. Figura 4

Figura 4 Estudio de hibridación in situ (FISH) con pérdida del gen señalada con sonda de color naranja para el gen SRY (Yp11.3).
Con base en la información previa se estableció el diagnóstico de síndrome De la Chapelle o síndrome del varón 46 XX. Se les propuso un tratamiento de infertilidad con donación seminal.
DISCUSIÓN
La manifestación clínica de este síndrome es variable y en la mayoría de los casos se trata de un fenotipo masculino normal; se clasifica en tres grupos conforme al fenotipo:14
El cuadro clínico se hará evidente, sobre todo en el primer grupo, después de la pubarca, con caracteres sexuales secundarios. Las manifestaciones clínicas más comunes son: ginecomastia, alteraciones en la longitud peneana, atrofia testicular, criptorquidia, hipospadias, infertilidad, azoospermia, ginecomastia, disminución de la masa muscular, alturas cortas y alopecia debida a la ausencia de testosterona.5,6 Los pacientes con este síndrome suelen recibir el diagnóstico durante la pubertad o en el proceso del estudio de infertilidad, por azoospermia e hipognadismo hipergonadotrópico.6,7,9 Estos datos clínicos ayudan a sospechar el síndrome y el diagnóstico final se establece con la ausencia del cromosoma Y en el cariotipo, que deberá corroborarse con la existencia o no del gen SRY.5
En las revisiones reportadas se informa que 83-89% de los pacientes con este síndrome tiene pérdida del gen SRY,4 entre 95 a 97% localizado en la porción Xp, lo que coincide con lo sucedido al paciente del caso. También se menciona que estos pacientes suelen ser de estatura baja, quizá debido a la traslocación de cromosomas sexuales que afectan la actividad hormonal del crecimiento, como sucedió en el paciente del caso.6,7
La mayoría de los casos se diagnostican durante la tercera década de la vida, con caracteres sexuales secundarios normales, con libido y erecciones, condiciones que tornan complejo el diagnóstico. El paciente del caso cumplió, prácticamente, con todos los criterios y las características mencionadas en la revisión. Es importante saber diferenciar las causas de hipogonadismo porque el tratamiento varía dependiendo de la causa que lo origina.6,7 El diagnóstico oportuno del síndrome permite iniciar la terapia de sustitución hormonal. El tratamiento oportuno evitará las complicaciones.
Antes de iniciar el tratamiento deben descartarse el cáncer de mama, el hematocrito mayor de 50%, apnea obstructiva del sueño, insuficiencia cardiaca o los síntomas urinarios bajos, que son contraindicación para recibir tratamiento con testosterona.6,15 La vía de administración, en gel transdérmico, intramuscular, parches o incluso en pellets no está estandarizada en estos pacientes.6 Antes de indicar la administración de testosterona convendrá practicar una densitometría ósea para descartar osteoporosis u osteopenia. En pacientes con T-score menor de -1 se sugiere indicar tratamiento con bisfosfonatos, vitamina D y calcio6 y continuar con seguimientos anuales. El tratamiento reproductivo deberá efectuarse con donación seminal con técnicas de alta o baja complejidad, dependiendo de los hallazgos durante el estudio de la pareja infértil.6
CONCLUSIONES
El síndrome del varón 46 XX es todo un reto para el clínico. Sus características dificultan el diagnóstico temprano debido a la evolución. Las ventajas de la terapia de remplazo hormonal, con testosterona, dependerán del tiempo de inicio que evite las consecuencias de su déficit. La ausencia de virilización, osteopenia o disminución de la libido no son las únicas complicaciones en estos pacientes. Es importante ayudarles a desarrollarse adecuadamente, dependiendo de la etapa de la vida en que se encuentren, buscando la estabilidad emocional y psicológica. Hay reportes en los que el diagnóstico ha sido prenatal, gracias a los hallazgos ultrasonográficos o por muestras de ADN fetal; sin embargo, la mayoría de los casos no permite iniciar la sustitución de manera temprana.
En pacientes con diagnóstico durante el estudio de infertilidad, la atención multidisciplinaria siempre será la ideal, como pudo hacerse en el paciente del caso. Al ser un síndrome que abarca 0.3% de los casos de infertilidad masculina y que el estudio genético no forma parte de manera rutinaria del algoritmo diagnóstico del hipogonadismo primario es importante recalcar que estos pacientes tienen que ser valorados por un especialista en Medicina de la Reproducción experimentado, con conocimientos sólidos de andrología. El médico debe conocer y dominar los algoritmos de estudio del hipogonadismo, saber practicar la adecuada exploración física masculina. Es importante saber explicar y hacer entender a la pareja estudiada que, debido a la azoospermia, es necesario recurrir a las técnicas de reproducción asistida, con semen de donante.

