










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de ingeniería biomédica
versión On-line ISSN 2395-9126versión impresa ISSN 0188-9532
Rev. mex. ing. bioméd vol.33 no.1 México abr. 2012
Artículo de investigación original
Modelos asociativos para la predicción de la localización subcelular de proteínas*
Associative models for the prediction of proteins subcellular localization
Acevedo-Mosqueda ME,* Acevedo-Mosqueda MA,* Calderón-Sambarino MJ**
* Escuela Superior de Ingeniería Mecánica y Eléctrica del IPN, Ciudad de México, México.
** Departamento de Ingeniería en Sistemas Computacionales de la Escuela Superior de Cómputo del IPN, Ciudad de México, México.
Correspondencia:
Acevedo-Mosqueda ME
Av. IPN s/n, Col. Lindavista 07738, Ciudad de México,
México, ESIME Zacatenco, Edif. Z-4, 3er Piso, Telecomunicaciones
Tel. 0155 57296000, Ext. 54757
-mail: eacevedo@ipn.mx
Artículo recibido: 18/octubre/2011
Artículo aceptado: 17/mayo/2012
Resumen
La localización de las proteínas dentro de la célula es fundamental para el entendimiento de su función biológica. Las proteínas son transportadas a orgánulos y suborgánulos específicos antes de ser sintetizadas. Son parte de la actividad celular y su función es eficiente cuando se encuentran en el lugar correcto. Es por esto que la localización de genes (codificados como proteínas) dentro de la célula se vuelve una tarea importante. En este trabajo se presenta un método para realizar la localización automática de genes dentro de la célula, como caso particular, se aplicó a la base de datos GENES. La propuesta tiene un enfoque asociativo y se utiliza, en particular, el modelo de las multimemorias asociativas alfa-beta. La efectividad en la localización obtenida fue del 97.99%, lo cual significa que este método, de 748 genes, no fue capaz de localizar sólo 14 de ellos.
Palabras clave: Predicción, localización subcelular, modelos asociativos, multimemorias alfa-beta.
Abstract
Protein subcellular localization is fundamental for understanding its biological function. Proteins are transported to specified cellular elements before they are synthesized. They are part of cellular activity and their function is efficient when they are in the right place. Therefore, genes (codified as proteins) localization into the cell becomes a key task. In this work, a method to localize automatically proteins into the cell is presented; as a particular case, the method was applied to the dataset GENES. The proposal has an associative approach and the specific model of alpha-beta associative multi-memories is applied. The effectiveness of the model was of 97.99%, which means that from 748 genes, the method was not able to localize 14 genes.
Key words: Prediction, subcellular localization, associative models, alpha-beta multimemories.
INTRODUCCIÓN
Las proteínas1 son las moléculas trabajadoras de una célula y llevan el programa de actividades, las cuales están codificadas por los genes. Dado que la estructura y función de una célula está determinada por las proteínas que contiene, el control de la expresión genética es un aspecto fundamental de la biología molecular de la célula. Las proteínas se pueden dividir en tres clases: globulares, fibrilares y de membrana. Comprender la función de las células requiere no sólo de la identificación de las proteínas expresadas en un tipo de célula dada, sino también de la caracterización de la localización de esas proteínas dentro de la célula.
Durante el proceso de la activación de la célula, las proteínas se dirigirán a los sitios subcelulares que les corresponden y jugarán su rol biológico2. Debido a que la localización es vital para la funcionalidad de la célula, las localizaciones que puede ocupar la proteína dentro de la célula conducen a diferentes efectos genéticos, tales como el cáncer u otras enfermedades genéticas.
Uno de los grandes retos que enfrentan los biólogos hoy en día es la clasificación funcional y estructural de las proteínas, así como también su caracterización3. Por ejemplo, en los humanos, el número de proteínas de las cuales se desconoce su función y su estructura es del 40% del total de proteínas, como resultado de esto, desde hace dos décadas se ha realizado una investigación muy extensa tratando de identificar las funciones y estructuras de las proteínas.
Por otro lado, a partir del conocimiento de las secuencias completas de un número de genomas4, el reto central de la bioinformática es la racionalización de toda la información obtenida de la secuencia, no sólo para encontrar estrategias más adecuadas para el almacenamiento de toda esta información, sino para diseñar herramientas de análisis más incisivas.
La inteligencia artificial proporciona esas herramientas permitiendo que el análisis sea más eficiente y efectivo. Es más eficiente en cuanto a que las decisiones respecto al análisis son más rápidas; y es más efectivo porque los resultados, la mayoría de las ocasiones, son más acertados que los resultados proporcionados por los expertos.
Un área de la inteligencia artificial son los sistemas expertos. Los recientes trabajos que se enfocan en la solución de problemas han dado como resultado el entender que la clave para obtener una solución es el conocimiento de un dominio en específico5. Por ejemplo, un doctor es efectivo diagnosticando enfermedades no porque tenga habilidades innatas para resolver problemas, sino porque tiene un vasto conocimiento del área. De manera similar, un geólogo es efectivo descubriendo depósitos minerales porque es capaz de aplicar un gran conjunto de conocimientos empíricos y teóricos acerca del área específica de la geología a la que se dedica. El conocimiento de un experto es la combinación de un entendimiento teórico de un problema en específico y la colección de reglas heurísticas para la solución de problemas que se obtienen mediante la experiencia. Por lo tanto, un sistema experto es construido con el conocimiento de un humano experto y codificándolo de forma que una computadora sea capaz de aplicarlo a problemas similares.
Una manera en que el experto humano elabora su diagnóstico es reconociendo las características principales de una enfermedad. Por ejemplo, el experto puede reconocer un tipo de arritmia con sólo visualizar un electrocardiograma (ECG), observando la amplitud o el ancho del complejo QRS. Un ortopedista puede detectar una fractura a partir de una radiografía porque sabe de antemano cómo luce la radiografía de una persona sana y cómo se ve una radiografía de un hueso fracturado. Esto es, el experto asocia las características que describen una anomalía con el diagnóstico de fractura y, de la misma manera, asocia las características presentadas por una radiografía de un hueso sano con el diagnóstico de una normal.
Este comportamiento de asociación natural que presentamos los seres humanos es simulado mediante una memoria asociativa. Por tanto, es posible que una memoria asociativa se utilice para tomar decisiones igual que hace un experto humano y comportase como un sistema experto.
El objetivo de este trabajo es utilizar un modelo asociativo que se comporte como un sistema experto decidiendo el lugar en donde se localizan las proteínas dependiendo de sus características.
Este trabajo está organizado de la siguiente manera: primero se describirá el problema planteado, a continuación se presentará la teoría de los modelos asociativos alfa-beta que es el fundamento de las multimemorias alfa-beta, posteriormente se describe la base de datos utilizada y los experimentos realizados junto con los resultados obtenidos. Finalmente, se presenta la sección de Discusión.
DESCRIPCIÓN DEL PROBLEMA
Debido a que la localización subcelular juega un papel importante en la función de la proteína, se vuelve esencial tener disponibles sistemas que puedan predecir la localización a partir de la secuencia para la completa caracterización de la expresión de las proteínas.
Se han utilizado algoritmos para la predicción subcelular6,7 de animales, plantas, hongos, de la bacteria Gram negativa8-11 y bacteria Gram positiva; estas predicciones se han hecho en forma separada o en conjunto12,13. También se ha hecho la predicción de proteínas procariotas y eucariotas14.
Hiyashi et al. utilizaron la base de datos GENES disponible en la página de KDD 200115, para realizar la tarea de localización de las proteínas. En ese trabajo se utilizó la técnica de los vecinos más cercanos (Nearest Neighbor) y obtuvieron una efectividad en la clasificación de 72.5%.
En este trabajo, se utilizó la misma base de datos para la tarea de localización. Se propone el uso de modelos asociativos alfa-beta. Esta propuesta se hace con base en la aplicación y en los resultados obtenidos por las diferentes memorias asociativas alfa-beta. Una de ellas es la memoria asociativa bidireccional alfa-beta16 que se ha aplicado como traductor inglés/español-español/inglés y en el reconocimiento de huellas digitales17, también se ha utilizado como clasificador de recurrencia de cáncer18 y para el almacenamiento de lattices de conceptos19. Algunas otras modificaciones al modelo original de la memoria asociativa alfa-beta son: redes neuronales alfa-beta20, memorias asociativas con soporte vectorial21 y difusas22, clasificador Gamma de alto desempeño23 y memoria asociativa para la selección de rasgos24.
En este caso, se aplicará el modelo de las multimemorias alfa-beta25 que ha demostrado ser una herramienta adecuada para la bioinformática26, ya que se ha aplicado en la clasificación de promotores de secuencias de ADN. El algoritmo se aplicó a dos bases estándar: E. coli y Primate splice-junction, los resultados de efectividad fueron del 98% y del 96.4%, en ambos casos este algoritmo arrojó los mejores resultados. La efectividad se comparó con métodos como la técnica de extracción M-of-N rules, Nearest-Neighbor, C4.5, Bayes y varios algoritmos basados en el perceptrón multicapa.
A continuación se describe el enfoque utilizado en este trabajo, desde los conceptos básicos hasta el modelo principal como lo son las multimemorias alfa-beta.
MODELOS ASOCIATIVOS
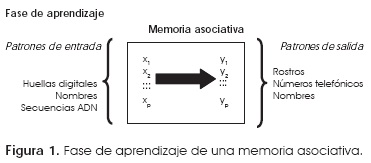
Una memoria asociativa (MA) es un sistema que relaciona patrones de entrada con patrones de salida. El objetivo de una memoria asociativa es recuperar el patrón de salida cuando se le presenta su patrón de entrada correspondiente.
El diseño de una MA necesita de dos fases: una de aprendizaje y una de recuperación. En la primera fase, la memoria debe ser capaz de asociar los patrones de entrada con los patrones de salida. En la fase de recuperación, la memoria asociativa recupera un patrón de salida correspondiente al patrón de entrada que recibe como información.
Si los patrones de entrada son diferentes a los patrones de salida, entonces la memoria asociativa es heteroasociativa. Por otro lado, si los patrones de entrada y salida son iguales, estamos hablando de una memoria autoasociativa.
Los patrones de entrada y salida pueden representar cualquier asociación, por ejemplo, huellas digitales asociadas a rostros, nombres con números telefónicos, secuencias de ADN con nombres, etcétera, esto se ilustra en la figura 1.
MEMORIAS ASOCIATIVAS ALFA-BETA
Las memorias asociativas alfa-beta27 utilizan el operador α en la fase de aprendizaje, y el operador β es usado en la fase de recuperación de patrones. Los conjuntos A y B, los cuales son: A = {0, 1} y B = {0, 1, 2} definen las operaciones binarias α y β mostradas en los cuadros 1 y 2, respectivamente.


Los conjuntos A y B, las operaciones binarias α y β junto con los operadores Λ (mínimo) y ν (máximo) usuales, conforman el sistema algebraico (A, B, α, β, Δ, V) en el que están inmersas las memorias asociativas alfa-beta. Dependiendo del operador utilizado en la fase de aprendizaje, mínimo o máximo, las MA alfa-beta pueden ser del tipo máx o mín, respectivamente.
MEMORIAS HETEROASOCIATIVAS ALFA-BETA
Una memoria es heteroasociativa si: yμ ≠ xμ ∀ μ ∈ {1, 2,..., p), donde μ es el índice que identifica a un número específico de patrón y p es el número de patrones asociados.
A continuación se definen las fases de aprendizaje y recuperación de una memoria heteroasociativa α-β.
Fase de aprendizaje
Paso 1. Para cada μ = 1, 2,..., p, a partir de la pareja (xμ,yμ) se construye la matriz: [xμ ⊗(yμ)t]mxn, donde m y n son la dimensión de los vectores x y y, respectivamente.
Paso 2. Se aplica el operador binario máximo v a las matrices obtenidas en el paso 1, si la memoria es de tipo máx, y si es de tipo mín entonces se aplica el operador binario mínimo A , como se muestra en las ecuaciones (1) y (2):
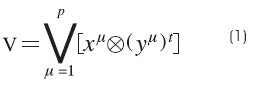
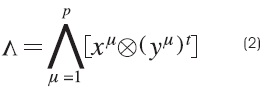
Fase de recuperación
El objetivo de esta fase es recuperar un patrón yω a partir de un patrón xω, cuando este último se le presenta a la memoria asociativa. El operador a utilizar en esta fase es β. Si en la fase de aprendizaje se creó una memoria de tipo máx, entonces el patrón xω operará con la matriz V junto con el operador mín ∧, por otro lado, si se aprendió con una memoria mín, entonces en la recuperación se operará el patrón xω con la memoria Λ utilizando el operador máx v , como se indica en la ecuaciones (3) y (4):


TIPOS DE RUIDO
Existen tres tipos de ruido a los que se enfrenta cualquier memoria asociativa que trabaja con valores binarios, los cuales son: aditivo, sustractivo y mezclado. Las memorias asociativas α-β máx y mín son capaces de manejar de manera adecuada el ruido aditivo y el ruido sustractivo, respectivamente. Sin embargo, ninguno de los dos tipos de memoria es adecuado para manejar el ruido mezclado (Cuadro 3).
Para tratar de evitar el ruido mezclado, el cual no puede ser manejado por ninguna de los dos tipos de memorias asociativas alfa-beta, se deben codificar los patrones a entrenar mediante el código Johnson-Möbius modificado.
CÓDIGO JOHNSON-MÖBIUS MODIFICADO
Las MA α-β máx son inmunes al ruido aditivo y las memorias mín al ruido sustractivo pero ninguna es capaz de manejar el ruido mezclado, es por eso que es necesario codificar los patrones antes de realizar la asociación. El código Johnson-Möbius modificado ha mostrado ser una buena opción para esto.
El código Johnson-Möbius es un código binario en el que sólo se permite el cambio de un bit al pasar de un número al siguiente28, y los bits cambiados permanecen hasta completar el ciclo y el cambio se inicia en el mismo extremo de cada sitio. Para representar un número decimal n en el código Johnson-Möbius, son necesarios n/2 bits si n es par, y (n + 1)/2 si n es impar.
A continuación se muestra un ejemplo utilizando el código original y el modificado.
Ejemplo de codificación Johnson-Möbius
Sea el conjunto r = {26, 2, 0, 5, 15} ⊂ R:
Paso 1. Como 26 es número par, entonces no es necesario sumarle 1.
Paso 2. Se divide rmáx/2= 13.
Paso 3. Si ri < rmáx/2, entonces se generan rmáx/2- ri ceros y se agregan ri unos. De lo contario, se generan rmáx-ri unos y se agregan ri - rmax/2 ceros.
El resultado se muestra en el cuadro IV.
Ejemplo de codificación Johnson-Möbius modificado29
Para el mismo conjunto de números anterior:
Para cada número ri del conjunto r, se generan rmáx-ri ceros concatenados con ri unos. El resultado se muestra en el cuadro 4.

Del cuadro 4 puede observarse que entre los patrones decimales 5 y 15 representados con el J-M modificado existe ruido aditivo si tomamos en cuenta como referencia el 5 ya que estaríamos cambiando ceros por unos para la representación del 15, o sería ruido sustractivo si ahora la referencia fuera el 15, ya que para la representación del 5 se estarían cambiando unos por ceros. Por otro lado, si la codificación utilizada fuera el J-M y tomando los mismo ejemplos anteriores, puede observarse que la representación del 15 decimal sería un patrón con ruido mezclado con respecto al patrón del 5 decimal.
De hecho, puede verse que cualquier patrón codificado con el J-M modificado, dentro del rango del 0 al 26 decimal, sería una versión con ruido aditivo o sustractivo de otro patrón que se encuentre dentro de ese mismo rango, ningún número en el rango establecido representará patrones con ruido mezclado. Por eso es necesario a la hora de codificar con el algoritmo J-M modificado, obtener los valores mínimos y máximos, de manera que cualquier número codificado sea un patrón con ruido aditivo o sustractivo y, por tanto, alguna de las dos memorias, máx o mín serán capaces de recuperarlos.
En el diseño de una MA, se crea una memoria por cada conjunto de patrones asociados. En el caso de una multimemoria, el conjunto de patrones asociados es dividido para crear varias memorias que finalmente operaran como una sola memoria. Antes de describir el diseño de la memoria se debe definir lo siguiente: Sean A = {0,1}, n, p Z+, μ {1, 2,... p}, i {1, 2,..., p} y j {1, 2,... n}, y sean x An y y Ap vectores de entrada y salida, respectivamente. A partir de estos vectores es posible construir el conjunto fundamental expresado por {(xμ, yμ)|μ = 1, 2,..., p} de acuerdo con el algoritmo de memoria heteroasociativa alfa-beta tipo mín.
MULTIMEMORIAS ALFA-BETA
Fase de aprendizaje
Para construir a partir de un solo conjunto fundamental varias matrices de aprendizaje, en lugar de sólo una, es necesario dividir cada patrón de entrada xμ en q particiones, generando así q vectores de entrada n/q-dimensionales y por lo tanto q memorias asociativas. Para cada μ {1, 2,..., p} a partir de la pareja ordenada (xμ, yμ) se aplica la operación de particionamiento vectorial a cada uno de los vectores xμ de las p parejas ordenadas.
Por lo que el nuevo conjunto fundamental es:
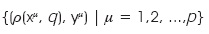
Aplicando el operador de particionamiento el conjunto fundamental queda como sigue:

y por distributividad:

Ahora, a partir de las parejas (Xμ1, yμ) se construyen las q matrices:
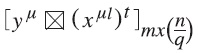
Se aplica el operador binario máximo a las matrices obtenidas si lo que se desea es construir una memoria máx, y se aplica el operador mínimo A si se va a construir una memoria mín, como se muestra en las ecuaciones (5) y (6), respectivamente:
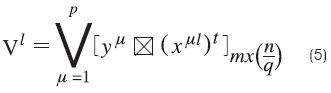

donde V l y Λl representan la l-ésima memoria máx y mín, respectivamente.
Fase de recuperación
Al igual que en la construcción de las matrices de aprendizaje, para la fase de recuperación es necesario dividir en q particiones el patrón que se presentará a las q memorias.
Paso 1. Al presentarse un patrón xω, con ω {1, 2,...p} a la multimemoria heteroasociativa alfa-beta, lo primero es aplicar el operador de particionamiento al vector xω De esta forma el vector quedará dividido en q nuevos vectores:
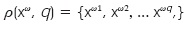
Ahora, para cada una de las q matrices y particiones del vector, se realiza la fase de recuperación del algoritmo de las memorias heteroasociativas alfa-beta tipo máx con la operación Δβ (ecuación (7)) y si la memoria es tipo mín se realiza con la operación ∇β como lo muestra la ecuación (8). Cualquiera de los dos resultados se asigna al vector zωl.


Una vez que se lleve a cabo este procedimiento se obtendrán q vectores resultantes, uno por cada una de las matrices de aprendizaje.
Paso 2. Se crea un vector intermedio el cual contendrá la suma de las i-ésimas componentes de los vectores zωl.
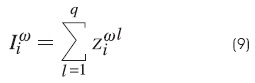
Una vez que se obtiene el vector el vector Iω, se obtendrá mediante la siguiente expresión:
Para la multimemoria máx:
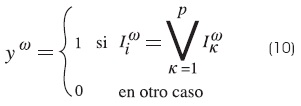
Para la multimemoria mín:
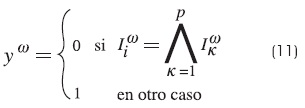
Multimemoria asociativa alfa-beta máx. Ejemplo.
Sean los 4 patrones de entrada, p = 4:
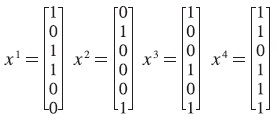
y sus correspondientes clases:
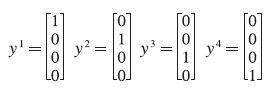
Fase de aprendizaje
Ahora se dividen los patrones de entrada en 3 particiones (q = 3) resultando 3 subvectores de dimensión 2, cada uno. Se aplica el operador de particionamiento vectorial p:
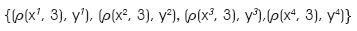
que también se puede expresar:
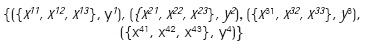
de manera particular cada partición queda como sigue:

por lo que el conjunto fundamental es el siguiente:
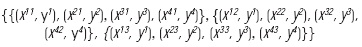
A partir de las parejas (Xμl, yμ), con μ = 1, ..., p y l = 1,..., q, se construyen las q matrices según la fase de aprendizaje. Por conveniencia, se construirá memoria asociativa máx, para el primer subconjunto de aprendizaje, {(x11, y1), (x21, y2), (x31, y3), (x41, y4)}:



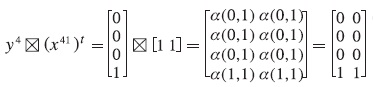
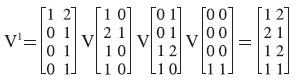
Se realiza el mismo procedimiento para obtener las otras dos matrices correspondientes a las memorias Máx, dando como resultado:

Fase de recuperación
Se le presenta el patrón x4 a las tres memorias asociativas máx construidas en la fase de aprendizaje.
En primer lugar, se le aplica el operador de particionamiento vectorial al patrón x4:
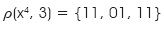
Ahora, cada uno de los subvectores es presentado a cada una de las tres memorias, como sigue:

Aplicando los mismos pasos, se obtienen los tres vectores z:
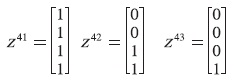
Se crea el vector intermedio utilizando la ecuación (9):

Debido a que se está trabajando con una memoria máx, se aplica la ecuación (10) para encontrar el patrón recuperado, tomando en cuenta que el valor máximo dentro del vector intermedio se encuentra en la cuarta componente:
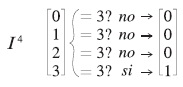
Por tanto:
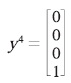
Se observa que se recupera el vector y4 que corresponde precisamente al vector asociado x4.
EXPERIMENTOS Y RESULTADOS
En esta sección se describirá la base de datos utilizada en este trabajo. Posteriormente, se describirán los experimentos realizados y, finalmente, se presentarán los resultados.
Base de datos GENES
Esta base de datos se encuentra disponible en la página de KDD 200115. El objetivo de este trabajo es la localización en la célula de un gen activo. Son 15 los lugares en donde se puede encontrar este gen y éstos son: pared celular, citoplasma, citoesqueleto, endosoma, ER, ambiente extracelular, Golgi, membrana integral, partícula grasa, mitocondria, núcleo, peroxisoma, membrana plasmática, vesícula de transporte y vacuola. En la figura 2 se muestra la posible localización de las proteínas (genes).
La base de datos contiene 862 registros para entrenamiento y 381 para prueba. Cada registro o gen tiene seis atributos: Essential, Class, Complex, Phenotype, Motif y Chromosome. Los valores que puede tomar Chromosome van de 1 a 16. Los otros cinco atributos están compuestos de diversos conjuntos. Por ejemplo, Class es un subconjunto de 24 categorías de proteínas, el atributo Complex indica los miembros de las 56 proteínas complejas codificadas por genes individuales. Phenotype y Motif asignan un gen a uno de los 11 fenotipos o 351 tipos, respectivamente.
La base de datos fue depurada debido a que algunos registros no tenían la información completa de los atributos, o los registros estaban repetidos o asignados a una misma clase (localización). El número de datos en el conjunto de entrenamiento fue de 784, mientras que el conjunto de prueba permaneció igual.
Experimentos
Cada uno de los seis atributos es codificado, por separado, mediante el código Johnson-Möbius. Después, se concatenan las codificaciones individuales para formar un solo vector que representará a cada registro. Esto se ilustra en la figura 3.

Una vez codificados todos los patrones de entrada, se eligen varios conjuntos de aprendizaje aleatoriamente. Del conjunto total de entrenamiento se tomó desde el 10% hasta el 100%, del conjunto de pruebas se tomaron todos los registros. Los diversos conjuntos de aprendizaje se muestran en el cuadro 5.
Con los diferentes conjuntos de aprendizaje se entrenan las multimemorias siguiendo el algoritmo de la fase de aprendizaje. Cada una de las memorias entrenada con cada conjunto de aprendizaje es probada con todos los registros del conjunto de prueba.
Para cada porcentaje se generaron, aleatoriamente, 100 conjuntos de aprendizaje y se probó con los 381 registros de prueba. Por cada una de las 100 pruebas se tomaron los 10 mejores resultados, los cuales se muestran en el cuadro 6.
Cuando se toma el 100% del conjunto de aprendizaje, el error que se obtiene es de 2.01, lo cual nos dice que la efectividad del modelo utilizado en este trabajo es de 97.99%. Esta efectividad nos indica que de 748 instancias, las multimemorias alfa-beta fallaron en reconocer sólo 14.
Hayashi et al., quienes utilizan la base de datos GENES, obtuvieron una efectividad del 72.5%. Nuestro modelo sobrepasó este resultado con un 25.49%. Del cuadro 6 podemos observar que un resultado similar es obtenido por las multimemorias alfa-beta cuando se utiliza el 30% del total de los registros de entrenamiento.
El cuadro 7 muestra los resultados obtenidos por algunos algoritmos de clasificación que se aplicaron a la base de datos GENES mediante la herramienta llamada WEKA30 versión 3.7.5. Este sistema contiene la implementación de varios clasificadores con distintos enfoques, por ejemplo: Bayes, regresión lineal, redes neuronales, basados en reglas, machine learning, entre otros. El formato original de la base de datos tuvo que ser modificado y convertido a mano al formato ARFF que maneja el software WEKA.

El cuadro 7 se puede verificar que el mejor resultado de efectividad, sin incluir nuestra propuesta, lo presenta el algoritmo que utiliza máquinas de soporte vectorial con el 90.34%. Una de las razones por las que estos enfoques tienen una baja efectividad puede deberse a la gran cantidad de rasgos que presenta cada registro en la base de datos. Por otra parte, la razón por la que nuestra propuesta presenta la mejor efectividad (97.99%) tiene su origen en la conjunción de dos sucesos: la codificación de los patrones mediante el código J-M modificado y el algoritmo de las multimemorias alfa-beta que han demostrado ser herramientas adecuadas para la clasificación de datos bioinformáticos.
En el cuadro 8 se pueden observar otros algoritmos de clasificación utilizados en la predicción de la localización de proteínas utilizando diferentes bases de datos.
La efectividad de estos enfoques va desde el 81% hasta el 97%. El enfoque propuesto en este trabajo, los modelos asociativos alfa-beta, presentan un 97.99% de efectividad superando en casi el 1% al algoritmo que presenta el mejor resultado.
La baja efectividad presentada por algunos de los algoritmos, de nuevo, es razonable debido a que las bases de datos utilizadas manejan grandes cantidades de información y el número de atributos es muy extenso lo que hace que la tarea de clasificación se vuelva compleja.
Cabe resaltar que, para este caso, no puede hacerse una comparación directa de los resultados presentados en este trabajo con los resultados mostrados en el cuadro 7, debido a que las bases de datos son diferentes y los algoritmos para la prueba de efectividad tampoco son los mismos.
Discusión
En general, los modelos asociativos representan una opción en al área de la inteligencia artificial, para las tareas de reconocimiento de patrones y clasificación. Los resultados obtenidos son comparables con algoritmos clásicos como las redes neuronales, y muchas veces son mejores. Sin embargo, los modelos asociativos alfa-beta presentan una «aparente» desventaja a la hora de su implementación. A continuación se explica la razón.
Las memorias alfa-beta tienen la capacidad de manejar grandes cantidades de ruido aditivo o sustractivo; esta tarea la realizan las memorias asociativas tipo máx y mín, respectivamente. Desafortunadamente, ninguno de los dos tipos de memoria puede manejar el ruido mezclado, sin importar si sólo se presenta un cambio de un 1 por un 0 y viceversa. Para tratar de disminuir la aparición de ruido mezclado en los patrones a reconocer o clasificar, utilizamos la codificación Johnson-Möbius modificada, la cual ha demostrado elevar la eficiencia de los modelos asociativos alfa-beta. En el cuadro 2 podemos apreciar que un patrón con un valor de 26 se representa con 26 bits. Ahora pensemos en un valor real de 9.234, por ejemplo, para que la memoria alfa-beta pudiera procesarlo sería necesario, primero, multiplicarlo por 1,000 para obtener el valor entero 9,234 y, posteriormente, se representaría con un vector de 9,234 elementos. Para el modelo presentado en este trabajo, tendríamos 18 multimemorias (9,234/18 = 513) construidas con patrones de 513 bits cada uno, lo que resultaría en memorias de 513 x p bits, donde p es el número de patrones asociados. Si p fuera igual a 1,000, entonces tendríamos 18 memorias de dimensión 513 x 1000. Ahora, si representamos cada 1 y 0 con arreglos del tipo carácter (8 bits), entonces las memorias ocuparían 18 x 513 x 1,000 = 9,234,000 bytes, lo que equivale a aproximadamente 9 MB de memoria. Otro tipo de memoria es la memoria asociativa bidireccional alfa-beta, en este caso, se tendría que construir una sola memoria con patrones de 9,234 + 1,000 = 10,234 elementos binarios, por tanto, la memoria tendría dimensiones de 10,234 x 10,234 lo que equivale a 104,734,756 ó 105 MB. Esta gran cantidad de memoria necesaria para almacenar el modelo asociativo es una «aparente» desventaja debido a que actualmente las computadoras disponen de memorias RAM muy grandes, además de procesadores muy potentes. Por otra parte, se pueden buscar algoritmos que puedan reducir los valores a codificar obteniendo así, una menor cantidad de elementos en los vectores. Por supuesto, existen otros tipos de modelos asociativos como los clásicos y los no clásicos como las memorias morfológicas que pueden manejar valores reales. Este tipo de memorias presentan las mismas desventajas que las alfa-beta en cuanto al manejo del ruido mezclado; sin embargo, utilizan otro tipo de métodos para la disminución de esta clase de ruido.
A pesar de la desventaja del almacenamiento del modelo asociativo en la memoria de la computadora, estos modelos siguen mostrando resultados comparables con las herramientas clásicas de inteligencia artificial.
CONCLUSIONES
Las multimemorias asociativas alfa-beta han encontrado su aplicación en el área de reconocimiento de patrones, incluida la tarea de clasificación. En particular, este modelo de memoria se ha aplicado en problemas relacionados con la bioinformática, obteniendo resultados favorables. Por lo tanto, en este trabajo se aplicó este modelo para la localización de proteínas dentro de la célula. Los resultados obtenidos fueron muy alentadores debido a que la efectividad fue del 97.99%, mientras que el mejor resultado obtenido por otros algoritmos fue del 97%. Cabe señalar que este resultado se obtuvo al aplicar nuestro modelo solamente a la base de datos GENES, aún no se ha aplicado a ninguna de las bases de datos que se muestran en el cuadro 8.
El siguiente paso es aplicar este modelo a las bases de datos que utilizaron los diversos algoritmos de clasificación que muestran en el cuadro 8. Además, se pretende usar este modelo utilizando otras bases de datos estándar que no pertenezcan a la bioinformática, esperando los mismos resultados favorables, sin embargo, debe recordarse el teorema del «no free lunch» que nos indica que un algoritmo puede ser excelente con ciertos datos y fallar con otro tipo de datos, lo que puede confirmarse en el caso de Hayashi et al, ya que ellos utilizan el algoritmo de 1-NN, el cual en la actualidad sigue siendo una de la herramientas más eficaces a la hora de clasificar, sin embargo, los resultados no fueron tan buenos.
AGRADECIMIENTOS
Los autores agradecemos la participación de Israel Román en la revisión del algoritmo.
REFERENCIAS
1. Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP et al. Molecular cell biology, 6th Edition, 2007: 59. [ Links ]
2. Juan EYT, Jhang JH, Li WJ. "Predicting protein subcellular localization using PsePSSM and suport vector machines". Proceedings of the 11th Join Conference on Information Sciences, 2008: 1-6. [ Links ]
3. Sarda D, Chua GH, Li KB, Krishnan A. "pSLIP: SVM based protein subcellular localization prediction using multiple physicochemical properties". BMC Bioinformatics 2005; 6: 152. [ Links ]
4. Feng ZP, "An overview on predicting the subcellular location of a protein". In: Silico Biology, 2002: 2. [ Links ]
5. Luger GF, Stubblefield WA. Artifical Intelligence: structures and strategies for complex problem solving. Addison-Wesley, Third-Edition, 1998: 20. [ Links ]
6. Yu-Dong C, Xiao-Jun L, Kuo-Chen C. "Artificial neural network model for predicting protein subcellular location". Computers and Chemistry 2002; 26: 179-182. [ Links ]
7. Yu CS, Chen YC, Lu CH, Hwang JK. "Prediction of protein subcellular localization". Proteins 2006; 64(3): 643-651. [ Links ]
8. Nakai K, Kanehisa M. "Expert system for predicting protein localization sites in Gram negative bacteria". Proteins 1991; 11(2): 95-110. [ Links ]
9. Gardy JL, Spencer C, Wang K, Ester M, Tusnády GE , Simon I et al. "PSORT-B: Improving protein subcellular localization prediction for Gram negative bacteria". Nucleic Acids Res 2003; 31(13): 3613-3617. [ Links ]
10. Yu CS, Lin CJ, Hwang JK. "Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions". Protein Sci 2004; 13(5): 1402-1406. [ Links ]
11. Lu Z, Szafron D, Greiner R, Lu P, Wishart DS, Poulin B et al. "Predicting subcellular localization of proteins using machine-learned classifier". Bioinformatics 2004; 20(4): 547-556. [ Links ]
12. Pierleoni A, Martelli PL, Fariselli P, Casadio R, "BaCelLo: a balanced subcellular localization predictor". Bioinformatics 2006; 22(14): e408-e416. [ Links ]
13. Chia-Yu E, Chiu H, Lo A, Hwang JK, Sung TY, Hsu WL. "Protein subcellular localization prediction based on compartment-specific features and structure conservation". BMC Bioinformatics 2007: 8-330. [ Links ]
14. Bing N, Yu-Huan J, Kai-Yan F, Wen-Cong L, Yu-Dong C, Guo-Zheng L. "Using AdaBoost for the prediction of subcellular location of prokaryotic and eukaryotic proteins". Mol Divers 2008; 12: 41-45. [ Links ]
15. Hayashi H, Sese J, Morishita S. Task: localization in KDD Cup 2001 report. Available: http://pages.cs.wisc.edu/~dpage/kddcup2001/ [ Links ]
16. Acevedo ME. Memorias asociativas bidireccionales alfa-beta. Tesis de Doctorado. Centro de Investigación en Computación, México, 2006. [ Links ]
17. Acevedo ME, Yáñez C, López I, "Alpha-beta bidirectional associative memories: theory and applications". Neural Processing Letters 2007; 26(1): 1-40. [ Links ]
18. Acevedo ME, Acevedo MA, Felipe F. "Classification of cancer recurrence with alpha-beta BAM". Hindawi Publishing Corporation 2009; 2009: 1-14. [ Links ]
19. Acevedo ME, Yáñez C, Acevedo MA. "Associative models for storing and retrieving concept lattices". Hindawi Publishing Corporation 2010; 2010: 1-26. [ Links ]
20. Argüelles A. "Redes neuronales Alfa-Beta sin pesos: teoría y factibilidad de implementación". Tesis de Doctorado. Centro de Investigación en Computación, México, 2007. [ Links ]
21. López L, "Máquinas asociativas alfa-beta con soporte vectorial". Tesis de Doctorado. Centro de Investigación en Computación, México, 2008. [ Links ]
22. Sánchez F. "Modelos Asociativos Alfa-Beta Difusos". Tesis de Doctorado. Centro de Investigación en Computación, México, 2009. [ Links ]
23. López I. "Clasificador automático de alto desempeño". Tesis de Maestría. Centro de Investigación en Computación, México, 2007. [ Links ]
24. Aldape M. "Enfoque asociativo para la selección de rasgos". Tesis de Doctorado. Centro de Investigación en Computación, México, 2011. [ Links ]
25. Román I, "Aplicación de los modelos asociativos alfa-beta a la bioinformática". MsD Thesis, Computing Research Center, Mexico City, Mexico, 2007. [ Links ]
26. Román I, López I, Yáñez-Márquez C. "Classifying patterns in bioinformatics databases by using alpha-beta associative memories". Stu in Comp Int 2009; 224: 187-210. [ Links ]
27. Yáñez C. "Memorias asociativas basadas en relaciones de orden y operadores binarios". PhD Thesis. Computing Research Center, Mexico City, Mexico, 2002. [ Links ]
28. Mano M. Diseño digital. Prentice-Hall, 2001: 16-26, 292-294. [ Links ]
29. Flores R. Memorias asociativas alfa-beta basadas en el código Johnson-Möbius modificado. MsD Thesis. Computing Research Center, Mexico City, Mexico, 2006. [ Links ]
30. Available: http://www.cs.waikato.ac.nz/ml/weka/
31. Reinhardt A, Hubbart T. "Using neural networks for prediction of the subcellular locations proteins". Nucleic Acids Research 1998; 6(9): 2230-2236. [ Links ]
32. Chou KC. "Prediction of protein subcellular locations by incorporating quasy-second-order effect". Biochemical Biophysical Res Commun 2000; 278: 477-483. [ Links ]
33. Wang J, Sung WK, Krishnan A, Li KB. "Protein subcellular localization prediction for Gram negative bacteria using aminoacid subalphabets and a combination of multiple support vector machines". BMC Bioinformatics 2005; 6(174): 1-10. [ Links ]
34. Tamura T, Akutsu T. "Subcellular localization predictions of proteins using support vector machines with alignment of block sequences utilizing amino acid composition". BMC Bioinformatics 2007; 8(466): 1-14. [ Links ]
35. Ogul H, Mumcuoglu EÜ. "Subcellular localization prediction with new protein encoding schemes". IEEE/ACM Trans Comp Bio and Biol Inf 2007; 4(2): 227-232. [ Links ]
36. Jin YH, Niu B, Feng KY, Lu WC, Cai YD, Li GZ. "Predicting subcellular localization with AdaBoost Learner". Protein Pept Lett 2008; 15(3): 286-289. [ Links ]
37. Xiao RQ, Guo YZ, Zeng YH, Tan HF, Pu XM, Li ML. "Using position specific scoring matrix and autocovariance to predict protein subnuclear localization". J Biomedical Science and Engineering, 2009; 2: 51-56. [ Links ]
38. Chou KC, Shen HB. "Plant-mPloc: A top-down strategy to augment the power" for predicting plant protein subcellular localization". PLoS ONE 2010; 5(6): 1-9. [ Links ]
Este trabajo fue apoyado por el Instituto Politécnico Nacional (COFAA, EDI y SIP) y el Sistema Nacional de Investigadores

