










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkHidrobiológica
versão impressa ISSN 0188-8897
Hidrobiológica vol.23 no.3 Ciudad de México Set./Dez. 2013
Artículos
La biodiversidad y evolución en ambientes acuáticos analizadas con herramientas moleculares
Biodiversity and evolution of aquatic environments analyzed by molecular tools
Alejandra Serrato Díaz1, Amelia Cornejo Romero2 y Odette Amilpa Castro3
1 Departamento de Hidrobiología. Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco 186, colonia Vicentina, 09340. México, D.F. México. E-mail: alej@xanum.uam.mx
2 Departamento de Biología. Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco 186, colonia Vicentina, 09340. México, D.F. México.
3 Maestría en Biología. Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco 186, colonia Vicentina, 09340. México, D.F. México.
Recibido: 12 de enero de 2012.
Aceptado: 18 de julio de 2013.
RESUMEN
Las herramientas moleculares constituyen un instrumento universal, sin precedentes, que ha revitalizado el estudio del origen y la evolución de la diversidad biológica de los complejos sistemas acuáticos. Estas herramientas, que incluyen marcadores moleculares y nuevos modelos teóricos y estadísticos para analizarlos, han ayudado a revelar patrones y procesos sobre biodiversidad, genética de poblaciones, filogeografía y evolución de los organismos que habitan estos sistemas, lo cual anteriormente no era posible hacer con las herramientas clásicas. Entre los avances obtenidos destacan: 1) el descubrimiento de una gran cantidad de especies, particularmente de microorganismos, y con ello el conocimiento de la complejidad y funcionalidad de las comunidades microbianas acuáticas, 2) la revelación de la estructura genética a escala fina, 3) la ampliación del conocimiento de las conductas filopátridas, la conexión entre poblaciones y la historia evolutiva de los linajes y 4) la descripción del origen, patrones de radiación, adaptación y coevolución a nivel macroevolutivo de diferentes taxones. El continuo mejoramiento de estas herramientas y la integración de otras disciplinas, ayudarán a tener una visión más completa sobre la biodiversidad, ecología y evolución en estos sistemas.
Palabras clave: Código de barras de la vida, estructura genética, filogeografía, macroevolución, metagenómica.
ABSTRACT
Molecular tools constitute an universal, unprecedented instrument to study the origin and evolution of biological diversity of complex aquatic systems. These tools, including molecular markers and new theoretical and statistical models to analyze them, have helped to reveal patterns and processes about biodiversity, population genetics, phylogeography and evolution of organisms that inhabit these systems, which previously was not possible with conventional tools. Advances included: 1) the discovery of a large number of species, particularly microorganism, and the knowledge of the complexity and functionality of aquatic microbial communities, 2) the unveiling of the genetic structure to fine-scale, 3) the extension of the knowledge about the phylopatry behavior, the connection between populations and the evolutionary history of gene lineages and 4) the description of the origin, radiation patterns, adaptation and coevolution at macroevolutive scale from different taxa. The continuous improvement of these tools and the integration of other disciplines help to have a more complete vision on biodiversity, ecology and evolution in these systems.
Key words: Barcode of life, genetic structure, macroevolution, metagenomics, phylogeography.
INTRODUCCIÓN
Los ambientes acuáticos concentran gran parte de la biodiversidad del planeta, lo que es comprensible si consideramos que la vida surgió en estos ambientes hace aproximadamente 3,500 millones de años, que comprenden más del 70% de la superficie de la Tierra y abarcan una gran variedad de hábitats, incluyendo dulceacuícolas, salobres y marinos (Jiménez et al., 2007). Sin embargo, la poca accesibilidad para muestrear y monitorear estos sistemas, debido a su extrema heterogeneidad espacial y temporal, dificultan el reto de comprender los procesos evolutivos que originan y mantienen su diversidad biológica (Selkoe et al., 2008).
En las últimas décadas, el desarrollo de nuevos marcadores moleculares, modelos teóricos y métodos estadísticos, así como la información genética disponible en bancos de datos y los equipos de cómputo de alta capacidad de procesamiento y almacenaje, han derivado en una explosión de estudios sobre biodiversidad y procesos evolutivos, en los que los ambientes acuáticos no han sido la excepción (Gilg & Hilbish, 2003; Schliewen & Klee, 2004; Zinger et al., 2012). Los avances logrados han ampliado nuestra comprensión sobre la diversidad y biología de los organismos acuáticos, han fortalecido las líneas de investigación tradicionales, han permitido plantear nuevas hipótesis y actualizar ideas predominantes en torno a la ecología y evolución (Schliewen & Klee, 2004; Selkoe et al., 2008). El objetivo del presente trabajo es ofrecer un panorama general sobre cómo las herramientas moleculares han revolucionado la manera de medir, estudiar y comprender la biodiversidad, ecología y evolución de la vida acuática.
DESCRIPCIÓN DE LA DIVERSIDAD BIOLÓGICA
Conocer la diversidad biológica es uno de los objetivos más importantes de la biología, sin embargo, estamos muy lejos de saber con exactitud cuántas especies habitan el planeta. Se estima que puede haber hasta cincuenta millones de especies, de las cuales solo se han inventariado un poco más de dos millones, aunado a este desconocimiento, frecuentemente nos enfrentamos a diferentes complicaciones para poder identificar a las especies que ya se conocen (May, 1988). Entre las dificultades para descubrir y distinguir a las especies se encuentran: la enorme diversidad del planeta, la presencia de un gran número de taxones unicelulares, los polimorfismos que presentan muchas especies a lo largo de su ciclo de vida, la existencia de especies con marcado dimorfismo sexual, la plasticidad fenotípica y las especies con convergencias morfológicas, así como la falta de taxónomos especialistas para la mayoría de los grupos de organismos (Stoeckle & Hebert, 2008).
Ante este panorama, las herramientas moleculares ofrecen una metodología de análisis uniforme que ayuda a descubrir, describir, categorizar y explicar la biodiversidad (Geller et al., 2010; Zinger et al., 2012). Como ejemplos de los alcances que tienen estas herramientas para estudiar la biodiversidad se encuentran el código de barras de la vida, utilizado para identificar a las especies (Stoeckle & Hebert, 2008; Pawlowski et al., 2012), y la metagenómica, que tiene como objetivo conocer la diversidad y el funcionamiento de una comunidad microbiana (Vogel & Nalin, 2003; Bonilla-Rosso et al., 2008; Lesk, 2008).
El código de barras consiste en identificar a las especies a partir de la secuencia de un fragmento estándar de ADN. Esta secuencia se puede obtener relativamente rápido a partir de especímenes de museos, herbarios, acuarios y tejidos congelados, entre otros (Stoeckle & Hebert, 2008). Hasta el momento, se han logrado estandarizar diferentes regiones de los genomas de un gran número de taxones como código de barras. La secuencia del gen mitocondrial citocromo oxidasa I (COI) se utiliza para identificar a la mayoría de los taxones animales, la secuencia de los genes de cloroplasto matK y rbcL para identificar algunos grupos de plantas y algas, y la región 18S rRNA para foraminíferos y diatomeas (Valentini et al., 2009). Con la aplicación de esta herramienta se han tenido avances impresionantes en el estudio de la biodiversidad. Se ha logrado: 1) identificar especies, como parásitos o invertebrados marinos, a lo largo de sus diversos estadíos larvarios (Resendiz, 2004), 2) documentar la diversidad biológica de taxones poco conocidos dentro de los grupos de plantas, hongos y animales microscópicos (Pawlowski et al., 2012), 3) estimar la biodiversidad e interacciones en una comunidad a partir de restos de los seres vivos que la componen (Yoccoz, 2012), 4) reconocer especies que aún no se han descrito y 5) diferenciar especies crípticas (Hebert et al., 2003; Witt et al., 2006; Valentini et al., 2009; Hajibabaei, 2012; Zhang & Hanner, 2012). Un ejemplo de la alta diversidad críptica que se ha encontrado en los ambientes acuáticos es el de los crustáceos del género Hyalella Smith 1874, que habitan en los cuerpos de agua del desierto de la Gran Cuenca en Norteamérica. En estos trabajos los autores encontraron que Hyalella sandra Baldinger, Shepard & Threloff 2000, son realmente dos especies crípticas y Hyalella azteca Saussure 1858, son 33 especies distintas, las cuales son imposibles de diferenciar morfológicamente (Hajibabaei et al., 2007; Stockle & Hebert, 2008). Sin embargo, donde ha tenido mayor impacto la aplicación de esta herramienta es en los microorganismos. En estos grupos, donde solo el 1% se puede cultivar y falta una delimitación morfológica clara, las secuencias de las regiones rpoB y 16S rRNA han permitido descubrir una cantidad inimaginable de nuevos Phyla de protistas y un acercamiento sin precedentes a las comunidades de arqueas y bacterias (Pawlowski et al., 2012; Zinger et al., 2012).
El código de barras de la vida es la primera herramienta que ofrece un criterio taxonómico universal, que se ha aplicado a un gran número de especies en todo el mundo, cuyos resultados se encuentran en una biblioteca pública de libre acceso (Hajibabaei, 2012). Sin embargo, esta novedosa herramienta tiene algunos problemas técnicos que es importante considerar. En primer lugar, no existe una región estandarizada para todos los grupos, particularmente ha sido difícil encontrar un código de barras en plantas (CBOL Plant Working Group, 2009), así como en otros taxones como los antozoarios y las esponjas, que presentan tasas evolutivas lentas para genes mitocondriales (Geller et al., 2010). En segundo lugar, la identificación y determinación de las especies dependen de los criterios taxonómicos de los especialistas de cada grupo, y cuando el taxón ha sido mal determinado, el código de barras nos dará una información incorrecta sobre esa especie. Por último, un pequeño fragmento del genoma no es un sustituto de todos los estudios biológicos que deben realizarse para conocer y entender a la biodiversidad (Will & Rubinoff, 2004).
Por su parte, la metagenómica es una disciplina que busca estudiar, desde diferentes puntos, a las comunidades microbianas (Vogel & Nalin, 2003; Bonilla-Rosso et al., 2008; Lesk, 2008). Consiste en extraer todo el ADN de muestras ambientales y secuenciar al azar un gran número de fragmentos. La finalidad es obtener el genoma completo de todos los microorganismos de ese ambiente, incluyendo el de los organismos de difícil aislamiento y cultivo, como lo son la mayoría de grupos de bacterias, arqueas y protozoarios (González et al., 2008). Mediante el análisis de algunas regiones de ADN, es posible inferir la historia evolutiva de cada una de las especies, explorar las rutas metabólicas, conocer los tipos de interacciones que mantienen las especies y rastrear la funcionalidad de las comunidades mediante el análisis de los genes (Vogel & Nalin, 2003; Bonilla-Rosso et al., 2008; Lesk, 2008).
Los fragmentos de ADN obtenidos, conocidos como reads, tienen un tamaño promedio de 500 pares de bases (pb). Los reads deben ensamblarse para armar el genoma de cada uno de los organismos. Para tener la secuencia precisa de una región determinada del genoma, se necesita que cada base, en cada sitio, haya sido secuenciada varias veces y que al alinear todos los fragmentos sean coherentes entre sí. A esto se le conoce como profundidad de la cobertura de secuenciación y se considera confiable si es mayor a 6x, lo cual implica que el total de las bases secuenciadas sea más de 6 veces el tamaño del genoma total (Bonilla-Rosso et al., 2008).
Un trabajo pionero en esta área es el desarrollado por Venter et al. (2004), en el cual se intentó obtener el genoma de las comunidades microbianas del Mar de los Sargazos. En dicho estudio se obtuvieron más de 1 billón de pb, en los que se detectaron más de 1.2 millones de genes y cerca de 1800 especies de microorganismos, de los cuales 148 correspondieron a nuevos registros en las bases de datos. Este proyecto arrojó descubrimientos muy importantes como el reconocimiento de la capacidad de los microorganismos para sintetizar nutrimentos a partir de componentes inorgánicos utilizando la luz solar como fuente de energía (fotoautotrofía) o la revelación de que los microorganismos marinos absorben principalmente la energía de la luz por medio de la rodopsina y no la clorofila como se había pensado (González et al., 2008). Se encontró una diversidad muy alta de proteorodopsinas, con al menos 782 homólogos, lo cual sugirió la existencia de un gremio diverso y abundante de fotoautótrofos adaptados de manera diferencial a nichos marinos para optimizar la absorción luminosa (Bonilla-Rosso et al., 2008).
El proyecto metagenómico más grande hasta ahora, es el Global Ocean Sampling (GOS) del J.C. Venter Institute. En éste se planteó muestrear 200 litros de agua cada 300 millas náuticas alrededor del mundo. En la primera parte del proyecto se obtuvo información sobre nuevas especies comparable con la obtenida en los viajes de colecta de Charles Darwin en el Beagle. Los resultados presentan el mayor aporte de secuencias nuevas y sin caracterizar a la base de datos del GenBank. Funcionalmente, se incorporaron a esta base de datos 6.12 millones de secuencias de proteínas, de las cuales un 23.4% representan familias de proteínas no descritas (Yooseph et al., 2007). Este estudio aportó información fundamental sobre la estructura y composición de las especies microbianas en las comunidades marinas de aguas superficiales, destacando que estas comunidades están dominadas por alrededor de 60 especies, de las cuales sólo una no había sido descrita, y que ninguno de los organismos dominantes pertenece al dominio Archaea, aunque previamente se había sugerido que este grupo dominaba los océanos.
En México se está desarrollando un estudio metagenómico en la localidad de Cuatro Ciénegas ubicada en el Desierto Chihuahuense en la región norte del país (Breitbart et al., 2009). Este sitio tiene los niveles más altos de endemismos en Norteamérica. Presenta una serie de pozas con gran diversidad de condiciones ambientales, más de 300 son cristalinas, algunas tienen temperaturas entre 30 y 40 °C y otras son ricas en sales. En ellas existe una amplia diversidad de comunidades formadoras de tapetes microbianos y estromatolitos. Los resultados obtenidos hasta ahora indican que la mayor parte de los microorganismos tienen un origen marino, que las comunidades son muy diversas, que presentan una alta diversidad metabólica, un alto endemismo y gran diferenciación ecológica debido a las características particulares de cada poza (Alcaraz et al., 2008; Bonilla-Rosso et al., 2008, 2012; Souza et al., 2008; Breitbart et al., 2009; Peimbert et al., 2012).
Los estudios de metagenómica realizados en los últimos dos años, han permitido encontrar una gran diversidad de especies de arqueas, bacterias y eucariontes unicelulares, incluyendo un gran porcentaje de especies nuevas. Esta información ha contribuido a definir los ecosistemas que habitan y conocer el potencial que tienen a nivel biotecnológico, industrial y médico, debido a que varias de esas especies pueden sintetizar gas o enzimas que proporcionan resistencia a altas temperaturas y salinidades (Somboonna et al., 2012). La metagenómica, sin lugar a dudas, ha aportado información invaluable sobre el mundo de los microorganismos y seguramente esta herramienta se seguirá aplicando a muchos ambientes para descubrir nuevos microorganismos, sus flujos genéticos, las redes metabólicas y las proteínas involucradas en éstas, a pesar del alto costo económico y de trabajo que ello implicará.
El código de barras de la vida y la metagenómica han modificado la manera de comprender la diversidad biológica, lo cual, transforma la manera de visualizar a las comunidades y tiene un importante impacto en muchas otras disciplinas como son: la sistemática, la conservación y la biotecnología, entre otras (Hajibabaei, 2012).
ESTRUCTURA GENÉTICA Y FILOGEOGRAFÍA
La diversidad biológica es producto de millones de años de divergencia y diversificación. La genética de poblaciones y la filogeografía son dos campos de estudio que se encargan de caracterizar la diversidad genética, determinar cómo se distribuye geográficamente y explicar los procesos ecológicos e históricos que la han generado (Avise, 2000; Beebee & Rowe, 2004; Hedrick, 2005). Estas disciplinas son de gran utilidad para los biólogos marinos interesados en caracterizar las escalas de la estructura genética y los patrones de conectividad (tasa de intercambio genético interpoblacional); revelar rutas de migración, conductas filopátridas y de apareamiento (Bowen & Karl, 2007); inferir procesos de especiación (Beheregaray, 2008); evaluar la capacidad de respuesta de las poblaciones y especies ante cambios ambientales naturales o antropogénicos e identificar poblaciones o especies que requieren acciones especiales de conservación (Selkoe et al., 2008; Geller et al., 2010).
Genética de poblaciones. El estudio de diversidad genética implica estimar la riqueza genética de las especies y determinar cómo se distribuye espacialmente. Con frecuencia las especies están compuestas por varias poblaciones que muestran diferencias genéticas a lo largo de su rango de distribución debido a la acción diferencial de la selección natural, deriva génica, mutación y flujo génico. Los primeros estudios de genética de poblaciones en especies marinas, basados en isoenzimas, mostraban que la estructura genética poblacional podía estar influenciada por el hábitat, la capacidad de dispersión y las conductas filopátridas, los cuales afectan el flujo génico (Ward et al., 1992; Bohonak, 1999). En especies con una alta capacidad de dispersión y/o amplia distribución se detectaron patrones de baja diferenciación genética interpoblacional (Avise, 2004). Así, en peces teleósteos marinos la baja diferenciación genética entre poblaciones se ha asociado al hecho de que los adultos llevan a cabo un gran movimiento, muchas veces vinculado con las corrientes marinas, y que en los océanos aparentemente en los océanos hay menos barreras al flujo génico (Ward et al., 1992). En las poblaciones de algunos peces del Pacífico con larvas pelágicas se manifiesta una uniformidad genética, contrario a lo observado en especies que carecen de esta fase (Avise, 2000).
También, se ha encontrado en algunas especies de invertebrados (erizos, langosta, camarón, mejillón) y peces (de arrecifes, costeros, cosmopolitas pelágicos perciformes y atún) que las fases larvarias planctónicas de larga duración ofrecen la oportunidad de llevar a cabo un intenso flujo génico a gran escala (Waples, 1987; Avise, 2004). Sin embargo, en otros organismos, como en la langosta americana, los copépodos y las lapas que presentan una alta capacidad de dispersión, se ha detectado a escala local una estructura llamada "parches genéticos caóticos". En dicha estructura, la variación entre sitios carece de una tendencia geográfica clara y/o muestra inestabilidad temporal, por lo que se dificulta establecer un patrón de diferenciación genética (Selkoe et al., 2010).
Debido a que las poblaciones marinas muestran patrones extremos de heterogeneidad temporal y espacial en factores demográficos como abundancia y tasas de migración que afectan la estructura espacial, es difícil detectar un patrón genético con base en los modelos de la genética de poblaciones clásica. Además, en muchos casos las especies no se ajustan a los supuestos teóricos de esta disciplina, como poblaciones panmíticas y discretas que intercambian migrantes solo entre las poblaciones vecinas más cercanas, por lo que se dificulta la explicación de las causas y patrones de la estructura genética (Selkoe et al., 2008).
La genética del paisaje ha surgido como una alternativa que mejora enormemente la comprensión de la estructura genética de poblaciones a pequeña escala y la conectividad. En esta disciplina emergente, la principal fuente de datos genéticos proviene de marcadores nucleares que son útiles para estudios a escala pequeña (local o regional) y a escala temporal más fina (Schoville et al., 2012). Los marcadores nucleares ampliamente empleados incluyen a los marcadores co-dominantes como los microsatélites, aunque es posible que éstos sean reemplazados en poco tiempo por los SNP's, debido a que facilitan la obtención de cientos o miles de datos a nivel de todo el genoma y los modelos de mutación son relativamente bien conocidos (Schoville et al., 2012).
El enfoque multidisciplinario de la genética del paisaje descansa en el análisis de la interacción entre los patrones genéticos y las características del paisaje, y explora las relaciones estadísticas entre la variación genética y la ambiental (Selkoe et al., 2010; Schoville et al., 2012). La aplicación de este novedoso enfoque ofrece la posibilidad de comprender mejor la estructura genética de especies marinas en las que la heterogeneidad ambiental y las herramientas clásicas de la genética de poblaciones no permitían establecer un patrón claro, como en los parches genéticos caóticos o en especies con poblacionales grandes en las que la divergencia genética es difícil de detectar.
Los modelos genéticos del paisaje marino emplean estimaciones empíricas de la composición y configuración de las características oceanográficas (corrientes marinas, barreras, gradientes, etc.); modelación espacial y datos de conducta para revelar patrones de la estructura genética, flujo génico y conectividad poblacional (Selkoe et al., 2008). Por ejemplo, se compararon las tasas de intercambio de larvas del mejillón azul (Mytilus edulis Linnaeus 1758) en una zona híbrida entre M. edulis y M. galloprovinicialis Linnaeus 1758 y la zona aledaña de la población de M. edulis, bajo diferentes patrones de circulación oceánica en Land's End, Inglaterra. Se llevó a cabo un muestreo bisemanal y se identificaron genéticamente a los reclutas puros e híbridos de M. edulis. Se determinó que el periodo, la distancia y tasas de dispersión coincidieron con el modelo hidrográfico (patrones de circulación oceánica), indicando que las larvas de M. edulis se comportan como partículas pasivas en las corrientes marinas superficiales. La distancia estimada promedio de dispersión fue de 30-50 Km, dependiendo de los patrones de circulación y la tasa anual de dispersión a través de la zona híbrida fue menor al 15%. Finalmente, los datos revelaron una proporción alta de retención natal, pues se encontró una correspondencia entre las áreas donde las larvas de M. edulis tenían una alta probabilidad de origen local con las áreas predichas por el modelo hidrográfico como zonas de alta retención (Gilg & Hilbish, 2003; Gilg et al., 2007).
La mayoría de los animales marinos producen larvas con un período de dispersión en la zona pelágica, el cual puede durar días o meses. La evidencia genética, oceanográfica y química de otolitos sugiere que al menos una proporción de larvas retorna a la misma subpoblación de sus padres (Selkoe et al., 2007; Planes et al., 2009). La tasa de intercambio genético entre poblaciones es un proceso que tiene efectos sobre el comportamiento y la dinámica de la población, la cual a su vez influencia la evolución, persistencia y resilencia de poblaciones de organismos marinos (Andras et al., 2013). Por lo que el conocimiento de la escala a la cual las poblaciones marinas están conectadas, a través de la dispersión de larvas, es crítico en la biología de la conservación, pues el grado de conectividad entre áreas geográficas determina la escala a la cual deben establecerse las redes de áreas marinas protegidas (MPAs, por sus siglas en inglés Marine Protected Areas) que son las principales herramientas utilizadas en planes de conservación de la biodiversidad y pesquerías marinas (Planes et al., 2009).
En el caso del róbalo marino, Paralabrax clathratus Girard 1854, se emplearon datos genéticos de varias series temporales de larvas, estructura de edades y la inferencia oceanográfica para examinar si el acervo de larvas de la población sureña del Canal de las islas de Santa Bárbara en California depende del suministro de larvas que se dispersan desde el norte de México, vía la corriente norte El Niño (Selkoe et al., 2007). Dado que los datos genéticos de diferenciación entre las poblaciones de los dos países fueron muy bajos, no se podía descartar su interdependencia. Sin embargo, los datos genéticos, complementados con la información ecológica y oceanográfica, sugieren que la variación interanual en el éxito del reclutamiento depende de factores locales y que la renovación del acervo de California y su manejo no depende de la dispersión de larvas provenientes de México (Selkoe et al., 2007).
Filogeografía. En esta disciplina se analiza cómo se distribuyen espacialmente los linajes de genes de una especie o especies cercanas para reconstruir su historia evolutiva (Avise et al., 1987; Avise, 2000). La filogeografía, surgida a finales de los años 70's, se basa en la teoría de la coalescencia para inferir en una genealogía de genes cómo convergen los linajes hacia su alelo ancestral y determinar si una secuencia es reciente o antigua. Las genealogías de genes, es decir, las filogenias de las distintas secuencias de ADN de un locus determinado de una especie, se construyen a partir de secuencias de ADN que se heredan uniparentalmente, como el mitocondrial (ADNmt), de cloroplasto (ADNcp) o genes nucleares de copia única y en algunos casos con microsatélites (Avise, 2009). El análisis filogeográfico permite poner a prueba hipótesis biogeográficas e inferir los procesos que subyacen al origen, distribución y mantenimiento de la biodiversidad (Beheregaray, 2008). También, permite estimar cambios en el tamaño histórico de la población e inferir cambios temporales y espaciales en el ambiente físico y biótico de las poblaciones (Avise, 1991; 2009). Lo anterior se basa en el hecho de que una población que ha mantenido un tamaño grande, preservará las distintas secuencias por más tiempo, pues la deriva génica no las extinguirá. De manera que los factores demográficos determinan los procesos de unión y ramificación de linajes. En un árbol de genes, dichos factores determinan la profundidad y forma de las ramas, pues en una población las hembras contribuyen diferencialmente a la progenie y cada línea materna, representada en las ramas, proliferará o desaparecerá (Figura 1a). Mientras que en las redes de haplotipos, el haplotipo más común, ubicado en la raíz de la red, representa la condición ancestral, además de que se distribuye ampliamente. Los haplotipos derivados y/o raros, ubicados en la periferia de la red, reflejan una expansión de su rango y tamaño poblacional conforme colonizaron nuevos ambientes (Figura 1b; Avise 2009).
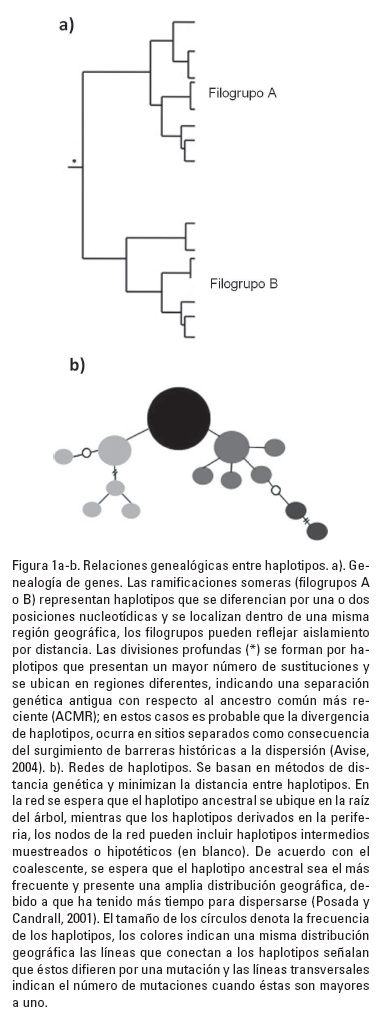
A partir de los estudios realizados hasta ahora, se han descubierto algunos patrones filogeográficos empíricos. Los peces de agua dulce frecuentemente muestran una marcada estructura filogeográfica que se asocia con cambios históricos y ecológicos de los ambientes acuáticos y del paisaje durante el Pleistoceno (Avise, 2000; 2009; Beheregaray, 2008). En contraste, los peces marinos comúnmente muestran un estructura filogeográfica débil, la cual se relaciona con la ausencia de barreras a la dispersión y altos niveles de conectividad espacial (Bowen & Grant, 1997). Asimismo, se ha incrementado nuestro conocimiento de la biología y evolución de diversos grupos con conductas filopátridas, como las tortugas y ballenas, y se ha ampliado nuestro conocimiento sobre los factores biogeográficos y ecológicos que moldean la distribución de su diversidad genética.
En la ballena jorobada (Megaptera novaeangliae Borowski 1781), que presenta filopatría y al mismo tiempo alta vagilidad, se ha detectado una estructura filogeográfica significativa. Las poblaciones dentro de cada una de las tres cuencas oceanográficas están subdivididas en matrilíneas de ADNmt que probablemente surgieron y son mantenidas por la fidelidad intergeneracional de las hembras a una ruta migratoria particular (Baker, 1993).
Las tortugas marinas (familias Cheloniidae y Dermochelyiidae) representan un grupo cuya estructura filogeográfica se ha estudiado ampliamente (Bowen & Karl, 2007). De acuerdo con estas investigaciones, se ha comprobado que las siete especies de tortugas marinas presentan fidelidad a sus sitios de anidamiento y que durante los estadíos juvenil y pre-adulto se mantienen en sitios de alimentación donde convergen tortugas de diferentes sitios de anidamiento. Asimismo, se ha demostrado que las tortugas presentan paternidad múltiple y se ha revelado un patrón filogeográfico global que muestra un gradiente determinado por la preferencia de hábitat y régimen térmico (Bowen & Karl, 2007). Los análisis de ADNmt y ADN nuclear (ADNnr) en estas tortugas muestran una estructura genética compleja. Por una parte, las secuencias de ADNmt revelan una fuerte estructura genética entre las colonias de anidamiento, es decir, las secuencias de cada región son únicas, no se comparten y corresponden al sitio de origen de las hembras. Por otra, los análisis de ADNnr muestran una estructura genética débil, resultado de un alto flujo génico mediado por los machos (Bowen & Karl, 2007).
Otro hallazgo importante es que los patrones de diferenciación genética han corroborado las divisiones biogeográficas propuestas para diferentes taxones, por ejemplo, el surgimiento del Istmo de Panamá, hace aproximadamente 3 millones de años, que separó el Océano Pacífico del Atlántico e interrumpió el flujo génico, afectando la distribución de las especies y su estructura genética. Se encontró que tanto en especies marinas, con bajo potencial de dispersión (un gran número de peces tropicales, camarones y erizos de mar), como aquellas con alta vagilidad (tortugas marinas), las genealogías de ADN-mt y otros genes arrojan árboles con ramificaciones profundas entre los linajes del Pacífico y el Atlántico reflejando la separación de estos océanos (Knowlton et al., 1993; Bermigham et al., 1997).
Los estudios de filogeografía comparada de especies co-distribuidas permiten ponderar la influencia de la dispersión y vicarianza e identificar las causas geológicas, ecológicas o etológicas que determinaron su evolución. Se ha detectado que en algunas especies con distribución ecológica similar, las estructuras genealógicas e historias demográficas son semejantes, sugiriendo que las fuerzas biogeográficas han moldeado las arquitecturas genéticas de biotas regionales (Avise, 2009). En varias poblaciones de peces dulceacuícolas de Norteamérica y Eurasia, vertebrados e invertebrados en la costa del Golfo de México y el Atlántico forman filogrupos, debido a que las poblaciones se contrajeron durante los cambios climáticos del Cuaternario (Avise, 1992; Bernardi et al., 1993; Bernatchez & Wilson, 1998). En catorce especies co-distribuidas de cuatro phyla (Echinodermata, Mollusca, Arthropoda y Chordata) en la zona rocosa intermareal del Pacífico en Norte América (desde Cape Flattery en Washington hasta la isla Kodiak en Alaska), se analizó la historia demográfica para determinar el efecto del último máximo glacial (UMG) sobre el tamaño efectivo (Ne) de las poblaciones. Se detectaron cambios en el Ne en 36% de las especies, lo cual indica una expansión demográfica poblacional después del UMG. Sin embargo, se observó que el 50% de las especies presentan Ne sin cambios a largo plazo, lo que indica que estas especies no fueron afectadas por el UMG (Marko et al., 2010).
MACROEVOLUCIÓN
Los estudios a nivel macroevolutivo integran toda la información paleontológica, embriológica, morfológica, genética y filogenética disponible para proponer un cuadro robusto de la evolución de los taxones superiores, con la finalidad de entender cuándo se originaron; investigar en qué momento surgen y cómo diversifican las características adaptativas; conocer los factores que limitan o favorecen las tasas y direcciones de la evolución; así como establecer cómo se relacionan unos grupos con otros (Hall, 2011). Dentro de las principales herramientas de los estudios macroevolutivos se encuentran las filogenias que son hipótesis acerca de la historia evolutiva de la vida. En las últimas décadas ha habido una enorme producción de filogenias moleculares que han revolucionado muchas hipótesis basadas en morfología y han proporcionado una nueva visión sobre la evolución de la vida (Sites et al., 2011). El uso de secuencias de ADN como caracteres ha revolucionado este campo debido, entre otras cosas, a que no tienen un efecto ambiental, la descripción de los estados de carácter no es ambigua, las regiones del genoma presentan diferentes tasas evolutivas de manera que se pueden hacer estudios en distintas escalas y a que algunos genes existen en todos los taxones por lo que se pueden incluir en un mismo estudio a grupos muy distantes que no comparten características morfológicas (Page & Holmes, 1998). Por otro lado, en las filogenias moleculares se puede estimar la edad de los grupos por medio del reloj molecular. Esta inferencia se basa en que un segmento de ADN acumula mutaciones a un ritmo lo suficientemente regular para medir hace cuánto tiempo se dio la divergencia entre dos taxones, tomando como referencia fechas obtenidas del registro fósil sobre el origen de los grupos que se están analizando (Page & Holmes, 1998; Hall, 2011). Aunado a esto, se han desarrollado poderosos métodos estadísticos de inferencia filogenética para analizar los datos y cada vez surgen más paquetes de análisis con mayor capacidad, a tal punto, que hoy en día ha surgido una nueva disciplina, la filogenómica, que consiste en la construcción de filogenias utilizando prácticamente el genoma completo de las especies (Ocaña & Dávila, 2011).
Una de las hipótesis que cambió drásticamente al incorporar en su estudio filogenias moleculares fue la de la clasificación de los seres vivos, cuando Woese & Fox (1977) propusieron un nuevo sistema de clasificación de tres dominios (nuevo nivel superior de clasificación) al comparar las secuencias de genes ribosomales de representantes de todos los reinos.
Las filogenias moleculares proporcionan un registro indirecto de los eventos que han dado origen a los grupos actuales por lo cual ofrecen un enorme potencial para estudiar diferentes procesos macroevolutivos como los que se mencionan a continuación.
Origen y evolución de caracteres. El conocimiento sobre el origen, la diversificación y el mantenimiento de los caracteres (morfológicos, fisiológicos, conductuales, etc.) permite proponer hipótesis sobre la historia adaptativa de los taxones (Sites et al., 2011). Para conseguirlo, los caracteres (incluyendo todos sus estados) deben ser mapeados sobre la filogenia calibrada del grupo que porta. A partir de la filogenia resultante se pueden inferir los grupos y el tiempo en que se originan, el estado ancestral, la historia de cambio y relacionar su origen con eventos geológicos, ecológicos o climáticos, para comprender la presión selectiva que favoreció su origen y mantenimiento, incluso cuando no existe información en el registro fósil (Sites et al., 2011). En la actualidad, con la aplicación de esta herramienta, se están cambiando las hipótesis sobre el origen y el mantenimiento de caracteres en diferentes niveles taxonómicos. Por ejemplo, Block et al. (1993), lograron proponer una hipótesis robusta sobre el origen de la endotermia (capacidad de mantener elevada la temperatura del cuerpo por medio del metabolismo) en el atún, caballas y peces espada del suborden Scombroidei. Esta característica, extremadamente rara en peces, fue mapeada sobre una filogenia molecular, dando como resultado que esta adaptación surgió de manera independiente en los tres linajes. Sin embargo, en los tres casos, se encuentra asociada con el movimiento de estos grupos hacia aguas frías. Este resultado permite inferir que es una característica adaptativa relacionada con la expansión de su nicho y genera nuevas preguntas como: ¿Qué originó el surgimiento en este grupo de esta rara característica por lo menos tres veces?
Origen y diversificación de los taxones. Durante mucho tiempo, el poder proponer cuándo y cómo surgieron los diferentes taxones, estuvo limitado al registro fósil que es escaso y cuando existe, no necesariamente representa el origen del taxón (Page & Holmes, 1998).
Como se mencionó, una herramienta poderosa para estimar la edad de los linajes y los tiempos de divergencia es la calibración del reloj molecular que proporciona información sobre el origen, tasas de diversificación y de extinción de los taxones. Esta herramienta se ha utilizado en el estudio evolutivo de innumerables grupos y ha cambiado muchos paradigmas sobre el tiempo en que éstos surgen y diversifican (Cooper & Penny, 1997; Page & Holmes, 1998). San Mauro et al. (2005), utilizaron esta herramienta para inferir la historia evolutiva de los anfibios. El origen y divergencia de este grupo es un tema que ha generado grandes debates debido a su pobre registro fósil. Con los resultados que estos autores obtuvieron, proponen que los tres órdenes de anfibios (Anura, Caudata y Gymnophiona), surgen en el Devónico de la era Paleozoica o principios del Mesozoico, antes de la separación de Pangea y poco después de la divergencia de los peces con aletas lobuladas, alrededor de 150 millones de años antes de lo que se suponía, modificando las hipótesis sobre el origen y evolución del grupo.
Radiación evolutiva. Existen numerosos taxones que presentan una gran riqueza de especies en comparación con sus grupos hermanos (Mayhew, 2006). Este fenómeno, conocido como radiación evolutiva o adaptativa, ha sido estudiado ampliamente bajo diferentes enfoques en una gran cantidad de grupos, sobre todo en los últimos años, debido al aumento de filogenias moleculares (Cristescu et al., 2010). La gran cantidad de información generada ha permitido proponer marcos conceptuales cada vez más completos e integrativos, que han llevado a definir y diagnosticar la radiación adaptativa, identificar los factores relacionados con este proceso y reconstruir la historia adaptativa de las radiaciones (Cristescu et al., 2010; Glor, 2010).
Los lagos antiguos del mundo como el Victoria, Malawi, Baikal, Caspio, Tanganica y Titicaca han sido excelentes sitios para estudiar la radiación, debido a que se tiene conocimiento sobre su historia geológica y contienen ejemplos clásicos de diversificación y especiación de un gran número de grupos acuícolas (Kocher, 2004; Cristescu et al., 2010; Glor, 2010). Los estudios en estos ambientes han permitido dilucidar los casos en los que si se trata de una radiación y han mostrado una gran diversidad de circunstancias que originan estas radiaciones, entre las que se pueden mencionar: 1) cuando se presenta un evento de colonización, el nuevo ambiente ofrece sitios disponibles que son ocupados por diferentes miembros del grupo y así inicia la rápida especiación o radiación; conforme se llenan los sitios y las especies se especializan, la tasa de especiación declina pues adquieren restricciones genéticas y fisiológicas que la limitan. Un ejemplo de este caso son los cíclidos del lago Victoria y de Malawi, así como los peces de la superfamilia Cottoidea del lago Baikal, en los tres casos existen evidencias filogenéticas de que la radiación se dio a partir de un solo ancestro colonizador, que se diversificó ante los nuevos espacios disponibles. 2) Se presentan múltiples colonizaciones con radiaciones independientes. Este patrón se encontró en la región Ponto-Caspio, donde ocurrieron colonizaciones múltiples de ancestros de crustáceos marinos y de agua dulce que dieron lugar a la radiación de cada uno de esos grupos. 3) Por efecto de la hibridación entre diferentes linajes alopátridos que se unen al colonizar nuevos ambientes. A pesar de que hace algunas décadas la hibridación no se consideraba un evento importante en la especiación, en los últimos años se ha demostrado que ha sido un factor clave en los eventos de radiación evolutiva de muchos taxones debido, entre otros aspectos, a que en las poblaciones híbridas aparecen características fenotípicas extremas que les permiten ocupar nuevos hábitats (Cristescu et al., 2010). Se ha reportado en diferentes taxones que los híbridos tienen características que les permiten colonizar nuevos sitios en los cuales se presenta radiación por los nuevos hábitats que se abren en la región colonizada. Otra consecuencia de la hibridación es que los alelos relacionados con la adaptación a determinados ambientes pueden llegar a otros grupos y mejorar, por ejemplo, la capacidad de los individuos que se encuentran en ríos para explorar o colonizar los hábitats de los lagos en los cuales se pueden presentar eventos de radiación (Schliewen & Klee, 2004).
Investigaciones en estos lagos, también han mostrado que muchos de los grupos propuestos como resultado de una radiación son en realidad resultado de múltiples eventos de colonización, algunas de las especies colonizadoras están preadaptadas a ciertos nichos más que divergir y especializarse en estos ambientes. En estos casos, se ha logrado descartar la posible radiación adaptativa debido a que los grupos no son monofiléticos lo cual es algo que caracteriza a la radiación (Day et al., 2007; Koblmüller et al., 2008).
Entre las investigaciones más relevantes sobre radiación evolutiva en los lagos antiguos se encuentra la de Brown et al. (2010), quienes estudiaron la historia evolutiva de la fauna, principalmente de anguilas, del lago Tanganica de África, uno de los más grandes del mundo que se caracteriza por su gran biodiversidad y endemismos. Estos autores realizaron un análisis filogenético molecular y encontraron que las anguilas colonizaron el lago hace 7 u 8 millones de años y tuvieron un evento de diversificación hace aproximadamente 2 ó 3 millones de años. Este evento coincide con el tiempo en el que los niveles del lago fueron muy bajos y la comunicación entre sus regiones disminuyó, causando el aislamiento de las poblaciones de diferentes especies, dicho evento geológico también coincide con la radiación de otros grupos de peces que habitan el mismo lago.
Finalmente, es importante mencionar que existen factores intrínsecos en los taxones que presentan esta diversificación repentina, debido a que no todos los grupos colonizadores de los lagos antiguos han presentado radiación evolutiva (Mayhew, 2006; Cristescu et al., 2010).
Coevolución. Todos los seres vivos mantienen diferentes tipos de interacciones que han sido clave en la generación y mantenimiento de la biodiversidad, debido a que proporcionan nuevos hábitats, afectan la abundancia y distribución de otras especies, dan estabilidad a los ecosistemas y que como resultado de lo anterior, tienen importantes consecuencias en la historia evolutiva de la vida (Jordano et al., 2009). En las últimas décadas, se ha abierto toda una nueva línea de investigación en el estudio de las interacciones al incorporar filogenias moleculares: la coevolución con un enfoque macroevolutivo. Algunas interacciones, como ciertos parasitismos, son muy específicas y antiguas, por lo que es posible proponer que las especies involucradas han compartido una larga historia evolutiva en la cual pudo ocurrir coevolución (evolución conjunta de dos o más taxones que no están relacionados filogenéticamente) (Ehrlich & Raven, 1964). Estudiar la evolución conjunta de estos grupos con filogenias morfológicas implica el problema de circularidad por las presiones selectivas generadas por la misma interacción. Sin embargo, al comparar las filogenias moleculares de los grupos implicados, es posible reconstruir su historia evolutiva y de esta manera inferir cuándo inició, bajo qué condiciones se estableció, sí ha existido oportunidad de evolución conjunta entre los grupos involucrados, sí existen cambios de hospedero, qué factores determinan esos cambios, así como establecer sí existen zonas de diversificación en la interacción (Brooks, 1988). Como resultado de esta comparación, se pueden obtener diferentes resultados, como la concordancia completa de ambas filogenias (Fig. 2a), donde una es el reflejo exacto de la otra. Este es un caso de coevolución estricta, producto de la especiación simultánea del hospedero y huésped (Page & Charleston, 1998). Sin embargo, las filogenias no siempre son completamente concordantes, debido a la presencia de otro tipo de eventos como la especiación independiente de uno de los grupos (Fig. 2b), la transferencia a otros hospederos, que pueden o no estar relacionados filogenéticamente al hospedero original (Fig. 2c), o bien la ausencia de un huésped en la descendencia de un hospedero que anteriormente lo tuvo (Fig. 2d) (Page, 1993; Page & Charleston, 1998; Clark et al., 2000; Roy, 2001). En este tipo de análisis, también es posible mapear sobre las filogenias caracteres importantes en la interacción y de esta manera estimar su valor adaptativo y su influencia en la diversificación y especificidad (Brooks, 1988; Page, 1993; Clark et al., 2000; Roy, 2001).

En las últimas dos décadas, se han realizado este tipo de estudios en diferentes sistemas, en los que existe una especificidad tan grande entre el hospedero y el huésped, que habían sido propuestos como ejemplos de coevolución estricta (Desdevises et al., 2002; Cernotíková et al., 2011). Los resultados obtenidos han permitido descubrir en estas interacciones un escenario dinámico y complejo, con especializaciones locales y mosaicos geográficos, con frecuentes cambios de hospedero y eventos de especiación independiente por parte de alguno de los taxones involucrados. Este patrón se ha encontrado en interacciones antagónicas y mutualismos y está generando el cambio de los modelos de coevolución estricta por escenarios complejos de coevolución difusa, donde interfieren diferentes factores ecológicos y evolutivos en la evolución de la interacción (Desdevises et al., 2002; Serrato, 2007).
CONSIDERACIONES FINALES
Las herramientas moleculares están revolucionando la manera de medir, estudiar y comprender la diversidad y evolución de los sistemas acuáticos. En la actualidad, estas herramientas permiten generar una gran cantidad de información molecular, incluyendo genomas completos, en unas cuantas horas. Esta nueva información, junto con la que está disponible en bancos de datos públicos, ofrece la oportunidad de resolver y plantear preguntas a un grado que no habíamos imaginado. La integración de esta información con datos demográficos, conductuales, biogeográficos, climáticos y sistemas de información geográfica, contribuirá a comprender los procesos involucrados en el origen y mantenimiento de la diversidad de estos ambientes.
Aunque sea relativamente accesible el uso de herramientas moleculares, es de vital importancia asegurar que su aplicación sea rigurosa y anclada en un cuerpo teórico que proporcione la comprensión sólida de los principios fundamentales sobre genética, biología evolutiva y ecología para evitar conclusiones y toma de decisiones equivocadas (Karl et al., 2012).
En países que albergan una gran diversidad en sistemas acuáticos y que han estado expuestos a un grave deterioro ambiental por causas antropogénicas, es necesario realizar un esfuerzo para aplicar estas herramientas y así tener una visión más completa sobre la biodiversidad y de esta manera proponer estrategias viables de conservación, manejo, protección y aprovechamiento de los recursos a diferentes escalas espaciales.
AGRADECIMIENTOS
Las autoras agradecen las observaciones y sugerencias de tres revisores anónimos que contribuyeron a mejorar el escrito.
REFERENCIAS
Alcaraz, L. D., G. Olmedo, G. Bonilla, R. Cerritos, G. Hernández, A. Cruz , E. Ramírez, C. Putonti, Β. Jiménez , E. Martínez, V. López, J. L. Arvizu, F. Ayala, F. Razo, J. Caballero, J. Siefert, L. Eguiarte, J. P. Vielle, O. Martínez, V. Souza, A. Herrera-Estrella & L. Herrera-Estrella. 2008. The genome of Bacillus coahuilensis reveals adaptations essential for survival in the relic of an ancient marine environment. Proceedings of the National Academy of Sciences of the United States of America 105: 5803-5808. [ Links ]
Andras, J. P., K. L. Rypien & C. D. Harvell. 2013. Range-wide population genetic structure of the Caribbean Sea fan coral, Gorgonia ventalina. Molecular Ecology 22: 56-73 [ Links ]
Avise, J. C., J. Arnold, R. M. Ball, E. Bermingham, T. Lamb, J. E. Neigel, C. A. Reeb & N. C. Saunders. 1987. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annual Reviews in Ecology and Systematics 18: 489-522. [ Links ]
Avise, J. C. 1991. Ten unorthodox perspectives on evolution prompted by comparative population genetic findings on mitochondrial DNA. Annual Reviews in Genetics 25: 45-69. [ Links ]
Avise, J. C. 1992. Molecular population structure and the biogeographic history of a regional fauna: A case history with lessons for conservation biology. Oikos 63: 62-76. [ Links ]
Avise, J. C. 2000. Phylogeography: The history and formation of species. Harvard University Press, Massachusetts, United States of America. 447 p. [ Links ]
Avise, J. C. 2004. Molecular markers, natural history and evolution. Sinauer Associates, Inc. Sunderland, United States of America. 511 p. [ Links ]
Avise, J. C. 2009. Phylogeography: retrospect and prospect. Journal of Biogeography 36: 3-15. [ Links ]
Baker, C. S. 1993. Abundant mitochondrial DNA variation and world-wide population structure in humpback whales. Proceedings of Natural Academy of Sciences 90: 8239-8243. [ Links ]
Beebee, T. J. C. & G. Rowe. 2004. An introduction to molecular ecology. Oxford University Press, New York United States of America. 346 p. [ Links ]
Beheregaray, Ι. Β. 2008. Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Molecular Ecology 17: 3754-3774. [ Links ]
Bermigham, E., S. S. McCafferty & A. P. Martin. 1997. Fish biogeography and molecular clocks: perspectives from the Panamanian Isthmus. In: Kocher, T. D. & C. A. Stepie (eds). Molecular systematic of fishes. Academic Press, San Diego, United States of America. pp 113-128. [ Links ]
Bernardi, G., P. Sordino & D. A. Powers. 1993. Concordant mitochondrial and nuclear DNA phylogenies for populations of the teleost fish Fundulus heteroclitus. Proceedings of the National Academy of Sciences USA 90: 9271-9274. [ Links ]
Bernatchez, I. & C. C. Wilson. 1998. Comparative phylogeography of Neartic and Paleartic fishes. Molecular Ecology 7: 431-452. [ Links ]
Block, Β. Α., J. R. Finnerty, A. F. Stewart & J. Kidd. 1993. Evolution of endothermy in fish: mapping physiological traits on a molecular phylogeny. Science 260: 210-214. [ Links ]
Bohonak, Α. J. 1999. Dispersal, gene flow and population structure. The Quarterly Review of Biology 74: 21-45. [ Links ]
Bonilla-Rosso, G., V. Souza & L. Eguiarte. 2008. Metagenómica, genómica y ecología molecular: la nueva ecología en el bicentenario de Darwin. Tip Revista Especializada en Ciencias Químico-Biológicas 11: 41-51. [ Links ]
Bonilla-Rosso, G., M. Peimbert, L. D. Alcaraz, I. Hernández, L. E. Eguiarte, G. Olmedo-Alvarez, & V. Souza. 2012. Comparative metagenomics of two microbial mats at Cuatro Ciénegas Basin II: Community structure and composition in oligotrophic environments. Astrobiology 12: 659-673. [ Links ]
Bowen, Β. W. & W. S. Grant. 1997. Phylogeography of the sardines (Sardinops spp.): assessing biogeograpic models and population histories in temperate upwelling zones. Evolution 51: 1601-1610. [ Links ]
Bowen, Β. W. & S. Α. Karl. 2007. Population genetics and phylogeography of sea turtles. Molecular Ecology 16: 4886-4907. [ Links ]
Breitbart, Μ., Α. Hoare, Α. Nitti, J. Siefert, M. Haynes, E. Dinsdale, R. Edwards, V. Souza, F. Rohwer & D. Hollander. 2009. Metagenomic and stable isotopic analyses of modern freshwater microbialites in Cuatro Ciénegas, Mexico. Environmental Microbiology 11: 16-34. [ Links ]
Brooks, D. R. 1988. Macroevolutionary comparisons of host and parasite phylogenies. Annual Review in Ecology and Systematics 19: 235-259. [ Links ]
Brown, K. J., L. Rüber, R. Bills & J. J. Day. 2010. Mastacembelid eels support Lake Tanganyika as an evolutionary hotspot of diversification. BMC Evolutionary Biology 19:188. doi:10.1186/1471-2148-10-188. [ Links ]
CBOL Plant Working Group. 2009. A DNA barcode for land plants. Proceedings of the National Academy of Sciences of the United States of America 107: 12794-12797. [ Links ]
Cernotíková, E., Α. Horák & F. Moravec. 2011. Phylogenetic relationships of some spirurine nematodes (Nematoda: Chromadorea: Rhabditida: Spirurina) parasitic in fishes inferred from SSU rRNA gene sequences. Folia Parasitologica 58: 135-148. [ Links ]
Clark, Μ. Α., Ν. Α. Moran, P. Baumann & J. J. Wernegreen. 2000. Cospeciation between bacterial endosymbionts (Buchnera) and a recent radiation of aphids (Uroleucon) and pitfalls of testing for phylogenetic congruence. Evolution 54: 517-525. [ Links ]
Cooper, Α. & D. Penny. 1997. Mass survival of birds across the Cretaceous-Tertiary boundary: molecular evidence. Science 5303: 1109-1113. [ Links ]
Cristescu, Μ. E., S. J. Adamowicz, J. J. Vaillant & D. G. Haffner. 2010. Ancient lakes revisited: from the ecology to the genetics of speciation. Molecular Ecology 19: 4837-4851. [ Links ]
Day, J. J., S. Santini & J. Garcia-Moreno. 2007. Phylogenetic relationships of the lake Tanganyika cichlid tribe Lamprologini: the story from mitochondrial DNA. Molecular Phylogenetics and Evolution 45: 629-42. [ Links ]
Desdevises, Y., S. Morand, O. Jousson & P. Legendre. 2002. Coevolution between Lamellodiscus (Monogenea: Diplectanidae) and Sparidae (Teleostei): The study of a complex host-parasite system. Evolution 56: 2459-2471. [ Links ]
Ehrlich, P. R. & P. H. Raven. 1964. Butterflies and Plants: A Study in Coevolution. Evolution 18: 586-608. [ Links ]
Geller, J. Β., J. Α. Darling & J. Carlton. 2010. Genetic perspectives on marine biological invasions. Annual Review of Marine Science 2: 367-393. [ Links ]
Gilg, Μ. R. & T. J. Hilbish. 2003. The geography of marine larval dispersal: coupling genetics with fine-scale physical oceanography. Ecology 84: 2989-2998. [ Links ]
Gilg, Μ. R., Kirby, S. E., Sullivan, R., Knapp, L. W. & T. J. Hilbish. 2007. Dispersal vs. retention: correspondence of species-specific reproductive cycles and settlement periods in a blue mussel hybrid zone. Marine Ecology Progress Series 351: 151-161. [ Links ]
Glor, R. E. 2010. Phylogenetic insights on adaptive radiation. The Annual Review of Ecology, Evolution, and Systematics 41: 251-270. [ Links ]
González, J. Μ., C. Pedrós-Alió & J. Μ. Gasol. 2008. Plancton bacteriano de los océanos. Investigación y Ciencia 387: 76-84. [ Links ]
Hajibabaei, Μ., G. Α. C. Singer, P. D. Ν. Hebert & D. Α. Hickey. 2007. DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends in genetics 23: 167-172. [ Links ]
Hajibabaei, Μ. 2012. The golden age of DNA metasystematics. Trends in genetics 28: 535-537. [ Links ]
Hall, Β. K. 2011. Evolution principles and processes. Jones and Bartlett. United States of America. 442 p. [ Links ]
Hebert, P. D. Ν., Α. Cywinska, S. L. Ball & J. R. Dewaard. 2003. Biological identifications through DNA barcodes. Proceedings of the Royal Society of London Series B-Biological Sciences 270: 313-321. [ Links ]
Hedrick, P. W. 2005. Genetics of Populations. 3a edition. Jones & Bartlett Learning, United States of America. 737 p. [ Links ]
Jiménez, J. C., Μ. Marfil, Α. Μ. Francesch, C. Cuevas, Μ. álvarez & F. Alberigo. 2007. Productos naturales de origen marino. Investigación y Ciencia 365: 75-83. [ Links ]
Jordano, P., D. Vázquez & J. Bascompte. 2009. Redes complejas de interacciones mutualistas planta-animal. In: Medel, R., M. A. Aizen & R. Zamora (Eds.). Ecología y evolución de interacciones planta-animal: conceptos y aplicaciones. Editorial Universitaria, S.A. Santiago de Chile, pp. 17-41. [ Links ]
Karl, S. Α., R. J. Toonen, W. S. Grant & Β. W. Bowen. 2012. Common misconceptions in molecular ecology: echoes of the modern synthesis. Molecular Ecology 17: 4171-4189. [ Links ]
Knowlton, Ν., L. Α. Weight, L. Α. Solórzano, D. Κ. Mills & E. Bermingham. 1993. Divergence of proteins, mitochondrial DNA, and reproductive compatibility across the Isthmus of Panama. Science 260: 1629-1632. [ Links ]
Koblmüller, S., Κ. Μ. Sefc & C. Sturmbauer. 2008. The Lake Tanganyika cichlid species assemblage: recent advances in molecular phylogenetics. Hydrobiologia 615: 5-20. [ Links ]
Kocher, T. D. 2004. Adaptive evolution and explosive speciation: the cichlid fish model. Nature Reviews Genetics 5: 288-298. [ Links ]
Lesk, Α. 2008. Introduction to Genomics. Oxford University Press. United King. 496 p. [ Links ]
Marko, P. Β., J. Μ. Hoffman, S. Α. Emme, T. Μ. McGovern, C. C. Keever & L. Ν. Cox. 2010. The "Expansion-Contraction" model of Pleistocene biogeography: rocky shores suffer a sea change? Molecular Ecology 19: 146-169. [ Links ]
May, R. Μ. 1988. How many species are there on earth? Science 247: 1441-1449. [ Links ]
Mayhew, P. J. 2006. Discovering evolutionary ecology. Bringing together ecology and evolution. Oxford University Press. New York. 212 p. [ Links ]
Ocaña, Κ. Α. & Α. Μ. Dávila. 2011. Phylogenomics-based reconstruction of protozoan species tree. Evolutionary Bioinformatic Online 7: 107-121. [ Links ]
Page, R. D. 1993. Genes, organisms and areas: The problem of multiple lineages. Systematic Biology 42: 77-84. [ Links ]
Page, R. D. & E. C. Holmes. 1998. Molecular Evolution: a Phylogenetic Approach. Blackwell Science. Oxford. 346 p. [ Links ]
Page, R. D. & Μ. Α. Charleston. 1998. Trees within trees: phylogeny and associations. Trends in Ecology and Evolution 13: 356-359. [ Links ]
Pawlowski, J., S. Audic, S. Adl, D. Bass, L. Belbahri, C. Berney, S. S. Bowser, I. Cepicka, J. Decelle, Μ. Dunthorn, Α. Μ. Fiore-Donno, G. H. Gile, Μ. Holzmann, R. Jahn, Μ. Jirku, P. J. Keeling, Μ. Kostka, Α. Kudryavtsev, E. Lara, J. Lukes, D. G. Mann, E. Α. Mitchell, F. Nitsche, Μ. Romeralo, G. W. Saunders, Α. G. Simpson, Α. V. Smirnov, J. L. Spouge, R. F. Stern, T. Stoeck, J. Zimmermann, D. Schindel & C. de Vargas. 2012. CBOL Protist Working Group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLOS Biology 10 (11): e1001419. [ Links ]
Peimbert, Μ., L. D. Alcaraz, G. Bonilla-Rosso, G. Olmedo-Álvarez, F. García-Oliva, L. Segovia, L. E. Eguiarte & V. Souza. 2012. Comparative meta-genomics of two microbial mats at Cuatro Ciénegas Basin I: Ancient lessons on how to cope with an environment under severe nutrient stress. Astrobiology 12: 648-658. [ Links ]
Planes, S., G. P. Jones & S. R. Thorroldd. 2009. Larval dispersal connects fish populations in a network of marine protected areas. Proceedings of the Natural Academy of Science 106: 5693-5697. [ Links ]
Posada, D. & Κ. Α. Crandall. 2001. Intraspecific gene genealogies: trees grafting into networks. Trends in Ecology and Evolution 16: 37-45. [ Links ]
Resendiz, Α. Ν. 2004. Descripción morfológica y caracterización molecular de la cercaria Glypthelmins quieta (Stafford, 1900) Stafford, 1905 (Platyhelminthes: Trematoda: Digenea). Tesis Licenciatura, Facultad de Ciencias, UNAM. México. 68 p. [ Links ]
Roy, Β. Α. 2001. Patterns of association between crucifers and their flower-mimic pathogens: Host jump are most common than coevolution or cospeciation. Evolution 53: 41-53. [ Links ]
San Mauro, D., Μ. Vences, Μ. Alcobendas, R. Zardoya & Α. Meyer. 2005. Initial diversification of living amphibians predated the breakup of Pangaea. The American Naturalist 165: 590-599. [ Links ]
Schliewen, υ. Κ. & Β. Klee. 2004. Reticulate sympatric speciation in Cameroonian Crater Lake cichlids. Frontiers in Zoology 1: 1-12. [ Links ]
Schoville, S. D., Α. Bonin, O. Francois, S. Lobreaux, C. Melodelima & S. Manel. 2012. Adaptive Genetic Variation on the Landscape: Methods and Cases. Annual Review of Ecology, Evolution and Systematics 43: 23-43. [ Links ]
Selkoe, Κ. Α., Α. Vogel & S. D. Gaines. 2007. Effects of ephemeral circulation on recruitment and connectivity of nearshore fish populations spanning the US-mexican border. Marine Ecology Progress Series 351: 209-220. [ Links ]
Selkoe, Κ. Α., C. Μ. Henzler & S. D. Gaines. 2008. Seascape genetics and the spatial ecology of marine populations. Fish and fisheries 9: 363-377. [ Links ]
Selkoe, Κ. Α., J. R.Watson, C. White, T. Β. Horin, Μ. Lacchei, S. Mitarai, D. Α. Siegel, S. D. Gaines & R. J. Toonen. 2010. Taking the chaos out of genetic patchiness: seascape genetics reveals ecological and oceanographic drivers of genetic patterns in three temperate reef species. Molecular Ecology 17: 3708-3726. [ Links ]
Serrato, Α. 2007. Coevolución a nivel macroevolutivo entre Ficus y sus polinizadores, las avispas Agaonidae en México. Tesis de Doctorado en Ciencias. CIEco, UNAM. México. 77 p. [ Links ]
Sites, J. W. Jr., T. W. Reeder & J. J. Wiens. 2011. Phylogenetics insights on evolutionary. Novelties in Lizards and snakes: sex, birth, bodies, niches, and venom. The Annual Review of Ecology, Evolution, and Systematics 42: 227-244. [ Links ]
Somboonna, Ν., Α. Assawamakin, Α. Wilantho, S. Tangphatsornruang & S. Tongsima. 2012. Metagenomic profiles of free-living archaea, bacteria and small eukaryotes in coastal areas of Sichang island, Thailand. BMC Genomics 13: S29. [ Links ]
Souza, V., L. E. Eguiarte, J. Siefert & J. J. Elser. 2008. Microbial endemism: does phosphorus limitation enhance speciation?. Nature Reviews Microbiology 6: 559-564. [ Links ]
Stockle, Μ. Y. & P. D. Ν Hebert. 2008. Barcode of Life. Scientific American 299: 82-88. [ Links ]
Valentini, Α., F. Pompanon & P. Taberlet. 2009. DNA barcoding for ecologists, Trends in Ecology and Evolution 24: 110-117. [ Links ]
Venter, J. C., Κ. Remington, J. F. Heidelberg, Α. L. Halpern, D. Rusch, J. Α. Eisen, D. Wu, I. Paulsen, Κ. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, Α. Η. Knap, Μ. W. Lomas, Κ. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, Η. Baden-Tillson, C., C. Pfannkoch, Y. Rogers & Η. 0. Smith. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304: 66-74. [ Links ]
Vogel, T. Μ. & R. Nalin. 2003. Sequencing the metagenome. ASM news 69: 107. [ Links ]
Waples, R. S. 1987. A multispecies approach to the analysis of gene flow in marine shore fishes. Evolution 41: 385-400. [ Links ]
Ward, R. D., D. O. F. Skibinski & M. Woodwark. 1992. Protein heterozygosity, protein structure, and taxonomic differentiation. Evolutionary Biology 26: 73-157. [ Links ]
Will, Κ. W. & D. Rubinoff. 2004. Myth of the molecule: DNA barcodes for species cannot replace morphology for identification and classification. Cladistics 20: 47-55. [ Links ]
Witt, J. D. S., D. L. Threloff & P. D. Ν. Hebert. 2006. DNA barcoding reveals extraordinary cryptic diversity in an amphipod genus: implications for desert spring conservation. Molecular Ecology 15: 3073-3082. [ Links ]
Woese, C. R. & G. E. Fox. 1977. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proceedings of the National Academy of Science of the United State of America 74: 5088-5090. [ Links ]
Yoccoz Ν. G. 2012. The future of environmental DNA in ecology. Molecular Ecology 21: 2031-2038. [ Links ]
Yooseph, S., G. Sutton, D. Β. Rusch, Α. L. Halpern, S. J. Williamson, Κ. Remington, J. Α. Eisen, Κ. Β. Heidelberg, G. Manning, W. Li, L. Jaroszewski, P. Cieplak, C. S. Miller, Η. Li, S. T. Mashiyama, Μ. P. Joachimiak, C. van Belle, J. Μ. Chandonia, D. Α. Soergel, Y. Zhai, Κ. Natarajan, S. Lee, Β. J. Raphael, V. Bafna, R. Friedman, S. E. Brenner, Α. Godzik, D. Eisenberg, J. E. Dixon, S. S. Taylor, R. L. Strausberg, Μ. Frazier & J. C. Venter. 2007. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biology 5:e16. [ Links ]
Zhang, J. & R. Hanner. 2012. Molecular approach to the identification of fish in the South China Sea. PLoS One 7 (2): e30621. [ Links ]
Zinger, L., Α. Gobet & T. Pommiers. 2012. Two decades of describing the unseen majority aquatic microbial diversity. Molecular Ecology 21: 1878-189. [ Links ]

