











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los canales de potasio de alta conductancia, dependientes de voltaje y de calcio (BK), se localizan próximos al receptor IP3 del retículo endoplásmico (IP3-R), formando un complejo (BK/IP3-R). Su función se relaciona con la excitabilidad de la neurona. Se promueve la hipótesis de que la hiperpolarización del potencial de membrana causada por la corriente BK (IBK) es un posible mecanismo capaz de suprimir la actividad eléctrica epiléptica en la neurona en algunos casos de epilepsia (Pérez-Bonilla et al., 2021).
Epilepsia
Aproximadamente 70 millones de personas en el mundo padecen de epilepsia (Thijs et al., 2019). Su etiología es múltiple (trastornos genéticos, isquémicos, traumáticos). Está relacionada con anormalidades en los circuitos neuronales (redes epilépticas) o anormalidades intrínsecas (neurona epiléptica) o ambas (Jefferys, 2010). Las características encefalográficas de la epilepsia se han estudiado ampliamente, y los aspectos biofísicos que subyacen se empiezan a conocer cada vez mejor (David et al., 2009). Actualmente, las investigaciones están dirigidas al estudio biofísico a dos niveles: redes neuronales (Fröhlich et al., 2010; Serrano-Reyes et al., 2020) y propiedades intrínsecas en neurona única (Araque et al., 2001; Steinlein, 2001). Sin embargo, los mecanismos a nivel celular de interrupción de la actividad epiléptica han sido escasamente abordados. Resultados teóricos, mediante modelos matemáticos computacionales, muestran que la terminación del tren de potenciales de acción (PA) asociado a convulsiones posictales es mediada por un incremento progresivo de sodio interno, seguido de una disminución de potasio externo (Krishnan & Bazhenov, 2011). En el presente trabajo se investiga un posible mecanismo intrínseco en la neurona para detener completamente o pausar temporalmente el tren de disparo relacionado con una actividad epiléptica.
Para este trabajo se utilizaron dos mecanismos epileptogénicos: (1) el incremento de la concentración de potasio extracelular ([K+]e) y (2) la mutación del canal de sodio dependiente de voltaje NaV 1.2. Dichos mecanismos epileptogénicos se implementaron computacionalmente para probar teóricamente, mediante simulaciones, el posible papel de protección del complejo BK/IP3-R en neuronas con actividad epiléptica.
Incremento de potasio extracelular en la génesis de epilepsia
Desde los años setenta se propuso la hipótesis de que algunos tipos de epilepsia están asociados a un incremento en la concentración externa de potasio ([K+]e). Fischer y colaboradores encontraron que un incremento de K+ en el espacio extracelular de 3.7 mM ± 0.6 mM a 6.9 mM ± 3.4 mM en corteza de hipocampo genera descargas interictales (Fisher et al., 1976). En la aplicación de 4-amino piridina (4-AP), el potasio extracelular se incrementó a 9 mM (Wang et al., 2016). La hipótesis de la acumulación de potasio externo ha retomado fuerza debido a los nuevos conocimientos de la dinámica de la concentración de los iones extracelulares (Fröhlich et al., 2008). En lesiones cerebrales que implican compromiso de la barrera hematoencefálica se han reportado focos epilépticos después de cuatro a siete días, debido a la disminución en la recaptura de glutamato y potasio a causa de una disfunción de los astrocitos (David et al., 2009).
La dinámica entre astrocitos y neuronas es una función crítica en el cerebro, se le asocia con plasticidad sináptica, modulación de la excitabilidad neuronal y balance iónico extracelular (Volman et al., 2012). El aumento de [K+]e favorece los mecanismos excitatorios en una red neuronal y la propagación de las ondas epileptiformes. La interacción neurona-glía ha sido abordada recientemente tanto experimentalmente como teóricamente (Chizhov & Sanin, 2020; Kager et al., 2000; Somjen et al., 2008).
Canalopatías asociadas a epilepsia
La epidemiología de la epilepsia está plagada de variaciones en la precisión del diagnóstico (Heron et al., 2007). Dentro de los pacientes con epilepsia, aproximadamente el 30% tiene una causa adquirida conocida, el restante 70% incluye a las epilepsias idiopáticas diagnosticadas electroclínicamente. En este grupo se distinguen como causas factores genéticos y mutaciones en los canales iónicos (canalopatías). La mayoría de las epilepsias idiopáticas en donde se conoce la base molecular son canalopatías (Campos-Castelló et al., 2002); sin embargo, es incierto el porcentaje de pacientes con epilepsia debido a una canalopatía (Heron et al., 2007). El aumento en la excitabilidad de las neuronas es una característica que está presente en cualquier síndrome epiléptico que se expresa como crisis convulsiva. Dentro de los mecanismos involucrados en este proceso se encuentran mutaciones en los genes de canales iónicos dependientes de voltaje o unidos a ligando (Campos-Castelló et al., 2002). Se ha reportado que muchas epilepsias causadas por canalopatías poseen un factor hereditario (Steinlein, 2001). En tales casos las mutaciones de los canales pueden heredarse de manera dominante o recesiva. Se inicia una nueva era con el reconocimiento de ciertos síndromes epilépticos como canalopatías y con cada vez una mejor comprensión de su fisiología molecular (George, 2004). Para Brunklaus y colaboradores es tiempo de ir más allá de la genética y encaminarse a una investigación genómica traslacional (Brunklaus et al., 2020). En la Tabla 1 se muestran las canalopatías asociadas a epilepsia genética, aunque son pocos los casos con este tipo de epilepsia (Steinlein, 2001). En este trabajo nos interesa la mutación del canal de sodio (NaV 1.2) en la génesis de epilepsia.
Tabla 1 Canalopatías asociadas a epilepsia genética.
| Canalopatías y epilepsia genética | |
| Gen/Cromosoma | Síndrome |
| Canales de potasio dependientes de voltaje | |
| KCNQ2, 20q13.2 | Convulsión familiar benigna tipo 1 |
| KCNQ3, 8q24 | Convulsión familiar benigna tipo 2 |
| SCN1B, 19q13.1 | Epilepsia generalizada con convulsiones febriles plus tipo 1 |
| SCN1A, 2q24 | Epilepsia generalizada con convulsiones febriles plus tipo 2 |
| Canales de sodio dependientes de voltaje | |
| SCN2A, 2q24 | Convulsión infantil-neonatal familiar benigna |
| CLCN2, 3q27.1 | Epilepsia mioclónica juvenil |
| Canales de calcio dependientes de voltaje | |
| CACNB4, 2q22-2q23 | Epilepsia mioclónica juvenil |
| CACNA1-A, 19q13 | Epilepsia generalizada con ataxia episódica |
| Receptor nicotínico (acetilcolina) | |
| CHRNA4, 20q13.2-q13.3 | Epilepsia autosómica dominante nocturna del lóbulo frontal tipo 1 |
| CHRNB2, 1q21 | Epilepsia autosómica dominante nocturna del lóbulo frontal tipo 3 |
| Receptor GABA* | |
| GABRA1, 5q34-q35 | Epilepsia mioclónica juvenil autosómica dominante |
| GABRg2, 5q31.1-q33.1 | Epilepsia generalizada con convulsiones febriles plus tipo 3 |
*ácido gamma amino butírico (GABA)
Fuente: Modificada de Steinlein (2001).
Mutaciones del canal NaV 1.2 en la génesis de epilepsia
Los canales de sodio dependientes de voltaje (NaV) están ligados a trastornos relacionados con la excitabilidad neuronal, dolor crónico y epilepsia. Existe una familia de canales selectivos a sodio formada por nueve subtipos (NaV 1.1 a NaV 1.9) (Eijkelkamp et al., 2012). Los canales NaV 1.1 y NaV 1.2 (genes SCN1A y SCN2A, respectivamente) son particularmente importantes en la generación de potenciales de acción. El subtipo de canal de sodio más relacionado con epilepsia es NaV 1.1 debido al gran número de mutaciones que producen epilepsia; se han reportado más de 1500 (Santos et al., 2020). En neuronas piramidales la mutación de NaV 1.2 produce un incremento en su función, generalmente asociada con convulsión neonatal infantil familiar benigna (BFNIS) (Kaplan et al., 2016). El registro en neuronas de animales B6.Q54 y F1.Q54 con mutaciones en NaV 1.2 presentan potenciales de acción espontáneos (Thompson et al., 2017). El ratón con mutación en NaV 1.2 (Scn2aQ54) presenta una epilepsia progresiva a causa de un aumento en la función del canal NaV 1.2 en el hipocampo (Kearney et al., 2001). Según el tipo de mutación del canal NaV 1.2, se puede producir un aumento o una disminución en su función. Los tipos de mutaciones en NaV 1.2 que aumentan la función del canal son los que interesan en el presente trabajo.
La función del complejo BK/IP3-R como un mecanismo de bloqueo de la actividad eléctrica convulsiva en neuronas
Los canales BK se activan por incremento de calcio interno. En el soma existen al menos dos fuentes de Ca2+ para activar al canal: (1) calcio que ingresa por los canales de Ca2+ dependientes de voltaje (CaV) localizados en la membrana celular (Grunnet & Kaufmann, 2004) y (2) calcio liberado de almacenes internos (Khodakhah & Ogden, 1995). Es bien conocida la interacción entre los canales BK/CaV. Se ha descrito un mecanismo de retroalimentación negativa entre los canales CaV y BK capaz de evitar la hipertensión arterial (Grimm & Sansom, 2010; Sachse et al., 2014). Se trata de un mecanismo de protección intrínseco de la célula. Sin embargo, el complejo BK/IP3-R se ha estudiado escasamente. El microdominio BK/CaV produce cambios en el potencial de membrana (PM), los cuales son relativamente fáciles de medir. El complejo BK/IP3-R no genera cambios del PM. Esto es una posible razón que explica los pocos trabajos reportados (Ross, 2012). La localización en distintas membranas de los canales BK y los receptores IP3 dificultan su estudio electrofisiológico. Se ha reportado (1) una cercanía y zonas de contacto entre la membrana celular y la membrana de retículo endoplásmico en la parte basal de las dendritas y el soma de neuronas (Wu et al., 2017), (2) la coexistencia BK/IP3 (Kaufmann et al., 2009) y (3) la liberación de calcio, en concentraciones de micromolar, desde el retículo endoplásmico (Larkum et al., 2003). Esto apoya la existencia de complejos BK/IP3-R en el soma. Un estudio teórico del microdominio BK/IP3-R sugiere una posible función como mecanismo de protección (Pérez-Bonilla et al., 2021). En neuronas se propone una triada CaV/BK/RyR-R (receptor a rianodina) como un posible mecanismo de protección (Irie & Trussell, 2017). Las preguntas son: ¿Existe algún mecanismo intrínseco en la neurona que suprima o trate de evitar la actividad eléctrica epiléptica o convulsiva en la neurona? ¿La función del complejo BK/IP3-R juega un papel como mecanismo intrínseco del cese de la actividad eléctrica epiléptica?
El objetivo de este trabajo es poner a prueba la siguiente hipótesis: En algunos tipos de epilepsia el complejo BK/IP3-R funciona como un mecanismo intrínseco de supresión de la actividad epiléptica neuronal. Si esto es así, entonces en un modelo computacional de epilepsia provocada en una neurona el complejo BK/IP3-R frena totalmente o pausa temporalmente los trenes de potenciales de acción epilépticos.
Materiales y métodos
La epilepsia es una condición disfuncional de las neuronas (factores intrínsecos) y redes neuronales (factores extrínsecos) (Jefferys, 2010). Los mecanismos asociados a la generación de epilepsia incluyen mutaciones en canales de sodio, calcio y potasio, disfunción de la homeostasis de iones extracelulares, cambios en la función de receptores y neurotrasmisores, y/o alteraciones en el balance excitación - inhibición en una red neuronal (McCormick & Contreras, 2001). En este trabajo, se plantea el siguiente supuesto: si una neurona silente es expuesta a uno de los mecanismos generadores de epilepsia (epileptogénesis) y produce trenes de potenciales de acción sostenidos sin ningún estímulo eléctrico, se considera que esta actividad eléctrica es epiléptica.
Para este propósito, se simuló una neurona con el formalismo de Hodgkin y Huxley (Hodgkin & Huxley, 1952). Esta neurona fue expuesta a dos mecanismos epileptogénicos: (1) incremento en la concentración de K+ extracelular y (2) mutación del canal de Na+ con incremento de su función.
Los modelos matemáticos utilizados en este trabajo son descritos parcialmente. En cada sección se hacen las citas correspondientes donde se encuentran los modelos matemáticos completos.
Modelo de neurona
El modelo matemático de Hodgkin & Huxley (1952) utilizado puede consultarse en Hodgkin & Huxley (1952), Pérez-Bonilla et al. (2021) y Sterratt et al. (2011). La ecuación expresa cuantitativamente el comportamiento de los canales de Na+, K+, BK y Cl- dependientes de voltaje (ecuación 1):
donde Cm es la capacitancia de membrana, V es el voltaje de membrana, INa es la corriente de Na+, IK es la corriente de K+, IL es una corriente de fuga e IBK es la corriente de K+ de alta conductancia dependiente de voltaje y de calcio.
Mecanismo de mutación del canal NaV 1.2
Para activar el mecanismo de generación de epilepsia por mutación del canal NaV 1.2, se modificó la cinética del canal de sodio en la ecuación 1. Se aumentó su conductancia y la duración de la inactivación (con mínima persistencia de la corriente), de acuerdo con el modelo de una corriente persistente de sodio (ecuación 2):
donde INaP es la corriente de sodio persistente, gNaP(max) es la conductancia máxima de sodio persistente, mNaP es la compuerta de activación, hNaP es la compuerta de inactivación, V es el voltaje de membrana y ENaP es el potencial de inversión para la corriente de sodio persistente (Krishnan & Bazhenov, 2011).
Mecanismo de incremento de K+ extracelular
Para reflejar el incremento de potasio extracelular en el potencial de membrana se utilizó el modelo de Goldman-Hodgkin-Katz, donde el voltaje de membrana está determinado por la ecuación 3:
donde V es voltaje de membrana; R es la constante de los gases; T es la temperatura; z es la valencia; F es la constante de Faraday; y PK, PNa y PCl son la permeabilidad a potasio, sodio y cloro, respectivamente. Los parámetros utilizados son: 1, 0.03 y 0.1 para PK, PNa y PCl, respectivamente. [K+], [Na+] y [Cl-] son las concentraciones de potasio, sodio y cloro, respectivamente, tanto fuera de la célula (e) como dentro (i). Los valores utilizados son (en mM): [K+]i = 110, [K+]e = 3.5 (modificable por el usuario), [Na+]i = 15, [Na+]e = 130 y [Cl-]i = 6, [Cl-]e = 130. Estos valores no cambian durante la simulación.
Los cambios de voltaje que se producen por variaciones del potencial de membrana por incremento [K+]e (Pm(K+)) se incorporan en la ecuación fenomenológica usada en este trabajo (ecuación 4):
donde Cm es la capacitancia de la membrana, V es voltaje de membrana y Pm(K+) es el potencial de membrana con cambios en K+ externo, calculado de la ecuación 3. gNa, gK, gBK y gl son las conductancias para el sodio, potasio, potasio de alta conductancia y de fuga, respectivamente. ENa, EK, EBK y El son los potenciales de equilibrio para sodio, potasio, potasio de alta conductancia y de fuga, respectivamente.
Modelaje del complejo BK/IP3
El sistema IP3
Para el modelado del Sistema IP3 se utilizó el modelo matemático de Goldbeter (1990). Relaciona dos variables principales: (1) la concentración de Ca2+ libre en el citosol (Z) y (2) la concentración de Ca2+ libre en el retículo citoplasmático (Y). Las ecuaciones 5 y 6 gobiernan la cinética de estas dos variables (Goldbeter et al., 1990; Pérez-Bonilla et al., 2021):
donde Z es [Ca2+] libre en el citosol, Y es [Ca2+] libre en el retículo citoplasmático, Vo es el flujo de calcio dentro de la célula, k es el flujo de calcio fuera de la célula, V1 es el flujo de calcio hacia el citosol desde el compartimiento sensible a IP3, b es la función de saturación entre el receptor e IP3, kf es la fuga de calcio del retículo citoplasmático al citosol, V2 expresa la velocidad de la bomba SERCA (Sarco(Endo)plasmic Reticulum Calcium transporting ATPase) y V3 es la velocidad de transporte del compartimiento sensible a calcio.
Este modelo genera oscilaciones de Ca2+ en el citosol, aumenta la concentración de calcio citosólico cuando existe una mayor liberación del retículo citoplásmico, por aumento de IP3 y/o de la velocidad de liberación y disminuye cuando aumenta la actividad de las bombas SERCA y PMCA (Plasma Membrane Ca2+ ATPase pump).
Modelo del canal BK
El canal BK fue modelado con la ecuación 7 (Pérez-Bonilla et al., 2021):
donde IBK es la corriente BK, gBK es la conductancia BK, q es la compuerta de inactivación, V es el voltaje de membrana y EK es el potencial de equilibrio para el potasio.
Con la implementación computacional de las ecuaciones se desarrollaron dos simuladores: (1) simulador “EpiNav” para probar la participación del sistema BK/IP3-R en la epilepsia generada por mutación de canal de Na+ 2.1 y (2) simulador “EpiKe” para probar el sistema BK/IP3 en la epilepsia generada por incremento de K+ extracelular.
El sistema de ecuaciones se resolvió simultáneamente por métodos numéricos (RungeKutta de cuarto orden, con dt = 0.01) y su función fue limitada para que los resultados se encuentren en valores fisiológicos.
Resultados
Se desarrollaron dos simuladores: (1) EpiNav: simulador interactivo que genera actividad eléctrica epiléptica en una neurona silente debido a la mutación del canal NaV 1.2 y (2) EpiKe: simulador interactivo que genera actividad eléctrica epiléptica en una neurona silente debido a un incremento de potasio extracelular.
Interfaz de usuario: módulos de datos comunes en los dos simuladores
Los dos simuladores cuentan con seis módulos (MOD) iguales para la entrada de datos (Figura 1): MOD I es el módulo de IP3 y gestión de oscilaciones de calcio intracelular que muestra las variables: n, m, p y r con valores iniciales de 2, 2, 4 y 4, respectivamente; MOD II es el módulo de salida de calcio hacia el citosol desde el almacén sensible a calcio, muestra la variable de fuga de calcio kf con valor inicial de 1 1/s; MOD III es el módulo de velocidad de salida de calcio por IP3, donde se encuentra la variable V3, relacionada con VM3, KR y KA con valores iniciales de 500 µM/s, 2 µM y 0.9 µM, respectivamente. MOD IV es el módulo para disminución de calcio intracelular por la bomba ATP (SERCA), bomba que ingresa calcio del citosol al almacén sensitivo a calcio, en este módulo se muestra las variables VM2 y K2 con valores iniciales de 65 µM/s y 1 µM, respectivamente; y MOD V es el módulo para la bomba PMCA que produce un eflujo de calcio hacia el espacio extracelular, aquí se muestra la variable K con valor inicial de 10 s-1. Los valores iniciales de estos cinco módulos derivan de Keener y Sneyd (Keener & Sneyd, 1998). MOD VI es el módulo que determina las concentraciones libres de calcio en el citoplasma y en el retículo con [Ca2+] = 0.1 µM y 100 µM, en el citosol y el retículo endoplásmico, respectivamente (Foskett et al., 2007). Estas condiciones propician una salida abrupta de Ca2+ del retículo, con una duración corta a manera de destello (spark) (Cheng & Lederer, 2008). Estos módulos corresponden a las variables que gobiernan el modelo de Goldbeter (1990). Finalmente, MOD VII es el módulo que determina la presencia de canales BK en la neurona, con variable gBK, con valores iniciales de 0 µS/cm2, condición control, sin canales BK (Figura 1).

Fuente: Elaboración propia.
Figura 1 Interfaz del simulador EpiNav. Se muestran los módulos de entrada de datos que controlan las oscilaciones de Ca2+ citosólico (MOD I al MOD VI). Los osciloscopios centrales registran estas oscilaciones: una salida abrupta de Ca2+ con una amplitud de ~5 µM seguida de oscilaciones de ~752 nM. En esta neurona no exciten canales BK, gBK = 0 mS/cm2 (MOD VII). El osciloscopio superior muestra el registro intracelular de la neurona (flecha roja), neurona silente. El osciloscopio inferior indica que no se estimuló eléctricamente a la neurona.
Módulos para datos específicos del simulador EpiNav y simulación de neurona silente
Este simulador cuenta con dos módulos específicos: (1) Módulo de pulso de estímulos de corriente, con una duración de 1500 ms y una amplitud inicial de 0 µA (sin estímulo eléctrico); y (2) módulo del canal NaV 1.2, con una conductancia inicial de 120 mS/cm2. Con estos valores iniciales se simula una neurona silente. El resultado de la simulación se presenta en la Figura 1. En el osciloscopio inferior no existe estímulo, amplitud = 0 nA. En osciloscopio en la parte media de la interfaz se despliegan las oscilaciones de Ca2+ intracelular. El osciloscopio superior muestra el registro eléctrico de la neurona, en este caso la neurona es silente.
Experimento generador de epilepsia por mutación del canal de Na+
La neurona silente anterior fue expuesta a una mutación del canal NaV 1.2, un mecanismo epileptogénico donde NaV 1.2 incrementa su función (Scalmani et al., 2006). Para este propósito se modificó la cinética del canal de sodio (constantes de tiempo y conductancia) en el modelo de Hodgkin & Huxley (1952). Desde la interfaz del simulador se puede modificar la conductancia de Na+ para incrementar su función. La Figura 2 muestra los resultados de la simulación al pasar la gNa de 120 mS/cm2 a 200 mS/cm2. Se observa en el osciloscopio superior cómo la neurona ahora dispara potenciales de acción de manera repetitiva y sostenida (actividad eléctrica epiléptica).

Fuente: Elaboración propia.
Figura 2 Simulación de epileptogénesis por mutación de NaV 1.2. A la neurona silente se le mutó el canal NaV 1.2. La flecha roja indica el incremento de la conductancia a 200 mS/cm2. La flecha azul muestra los disparos de potenciales de acción repetitivos y sostenidos (neurona epiléptica). Las demás variables permanecen con valores iguales a la neurona anterior.
Efecto del complejo BK/IP3-R en la epilepsia por mutación del canal de Na+
Se propone que el complejo BK/IP3-R es un sistema intrínseco que juega un papel importante en la finalización de la actividad eléctrica epiléptica de la neurona. Si esto es así, entonces deberá detener el estado epiléptico. Se realizó el siguiente experimento virtual. A la neurona epiléptica anterior se le mantuvo el incremento de gNa = 200 mS/cm2 con la finalidad de continuar con el estado epiléptico. La Figura 3 muestra dos simulaciones superpuestas: (1) la simulación donde se generó epilepsia (tren de PA en el osciloscopio superior, por mutación de NaV 1.2, gNa = 200 mS/cm2, gBK = 0 µS/cm2) y (2) la simulación donde se agregaron canales BK a la neurona (gBK = 0.6 µS/cm2); el complejo BK/IP3-R ahora está completo y activo. En esta última condición, se puede observar cómo la salida abrupta de calcio (flecha roja en el osciloscopio central) activa a los canales BK (gBK = 0.6, circulo en rojo). Como resultado, se detiene la actividad eléctrica epiléptica de la neurona (trazo en rojo en el osciloscopio superior, señalado por la flecha). Todas las demás variables mantienen los valores de la neurona epiléptica anterior.
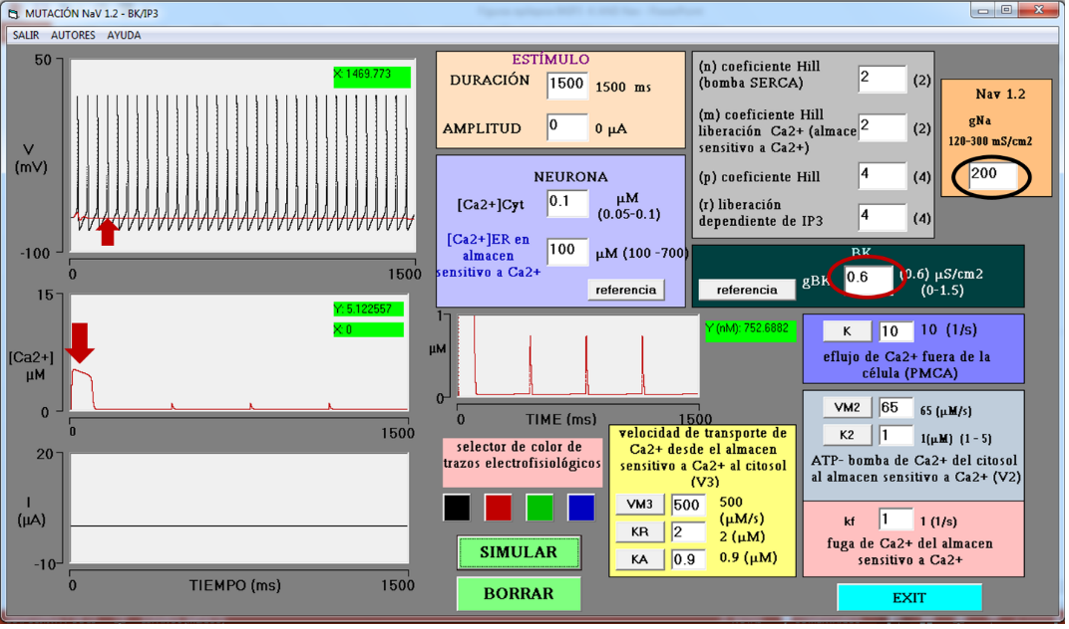
Fuente: Elaboración propia.
Figura 3 Efecto del complejo BK/IP3-R en la epilepsia simulada por mutación del canal de sodio. En el osciloscopio central se observa la salida de Ca2+ del retículo y sus oscilaciones. La amplitud de la salida abrupta de Ca2+ alcanza 5.12 mM (flecha roja inferior). La conductancia de sodio permanece en 200 mS/cm2 (círculo negro). La conductancia gBK es de 0.6 µS/cm2 (círculo rojo). La activación de los canales BK por Ca2+ bloquea completamente la actividad eléctrica epiléptica de la neurona (trazo en rojo, señalado con flecha roja, en el osciloscopio superior).
Simulador EpiKe
Interfaz de usuario
Este simulador cuenta con un módulo de entrada de datos específico, donde se puede aumentar la concentración de potasio externo y el tiempo de simulación. Los valores iniciales para estas variables al ejecutarse el simulador son: [K+]e = 3.5 mM (círculo negro) y un tiempo de simulación de 3500 ms. Las demás variables permanecen con valores iniciales descritos arriba. En estas condiciones la neurona no dispara potenciales de acción, es silente (Figura 4, flecha negra).

Experimento de epileptogénesis por incremento [K+]e
La concentración de potasio externo incrementa la excitabilidad en la neurona y puede causar actividad neuronal espontánea, mecanismo encontrado en situaciones anómalas como epilepsia (Traynelis & Dingledine, 1988). Concentraciones altas de potasio externo, debido a disfunción de los astrocitos, son capaces de generar focos epilépticos (David et al., 2009). En las condiciones de concentraciones utilizadas en el modelo matemático se realizaron varios experimentos virtuales, se probaron varias [K+]e hasta lograr que una neurona silente generara un tren de potenciales de acción sostenidos (epileptogénesis por K+ externo), el resto de las variables permaneció sin cambio. La Figura 5 muestra la simulación con [K+]e = 10 mM (círculo rojo). Con esta concentración se logró que una neurona silente tenga actividad epiléptica (flecha roja). Esta concentración de K+ externo es similar a las reportadas en sacudidas epilépticas donde se tienen oscilaciones de [K+]e entre 15 mM y 20 mM (Chizhov & Sanin, 2020). El valor de las demás variables se mantuvo sin cambio.
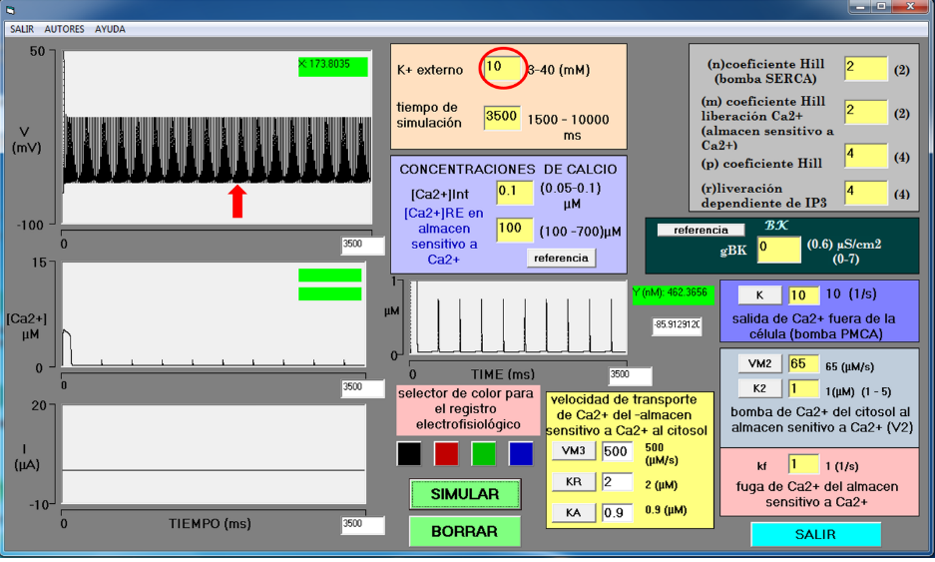
Fuente: Elaboración propia.
Figura 5 Proceso epileptogénico por aumento de potasio externo. La concentración de K+ externo se aumentó de 3.5 mM a 10 mM (círculo rojo). En el osciloscopio superior se muestra la actividad epiléptica generada (trazo negro, flecha roja). Los valores de las demás variables permanecen sin modificaciones.
Efecto parcial del complejo BK/IP3-R en la epilepsia por incremento [K+]e
Los canales BK activados por incremento en [Ca2+]i y voltaje producen una poshiperpolarización rápida (Bock & Stuart, 2016). Esta actividad es contraria a la despolarización sostenida causada por el incremento en la concentración de potasio externo (Chizhov & Sanin, 2020). La Figura 6 muestra la superposición de dos registros: (1) Registro en neurona con actividad eléctrica epiléptica por [K+]e = 10 mM (trazo en negro), resto de las variables sin cambio, y (2) Registro en la misma neurona al activarse el complejo BK/IP3-R por aumento de gBK de 0 µS/cm2 a 6 µS/cm2 (círculo rojo) y de VM3 de 500 a 850 (µM/s) (círculo azul) para incrementar la concentración de calcio interno (flecha azul). El resultado fue una pausa del tren de potenciales de acción debida a la poshiperpolarización causada por la actividad de los canales BK (Figura 6, trazo en rojo, flecha roja). Sin embargo, la hiperpolarización del complejo BK/IP3-R y la despolarización causada por aumento sostenido de [K+]e están en competencia. Después de la poshiperpolarización, la neurona inicia una despolarización lenta y constante hasta que, en este caso, a los 1340 ms, la neurona nuevamente genera potenciales de acción sostenidos. El complejo BK/IP3-R no fue capaz de frenar totalmente el estado epiléptico, pero sí de suspenderlo temporalmente. Como la actividad epiléptica está relacionada con la sincronización de una red neuronal (Serrano-Reyes et al., 2020), una suspensión temporal en varias neuronas debido a la actividad del complejo BK/IP3-R podría desincronizar a la red, al menos temporalmente, y pausar la actividad eléctrica epiléptica. La hiperpolarización causada por los canales BK no frena inmediatamente al tren de potenciales de acción y se produce una ráfaga de PA inicial, seguido de una pausa, para después nuevamente producir potenciales de acción sostenidos. En caso de que el complejo BK/IP3-R se active frecuentemente para intentar suprimir el estado epiléptico, entonces se producirá una actividad neuronal combinada entre ráfagas de potenciales de acción y potenciales de acción sostenidos, actividad que recuerda a un estado tónico-clónico.
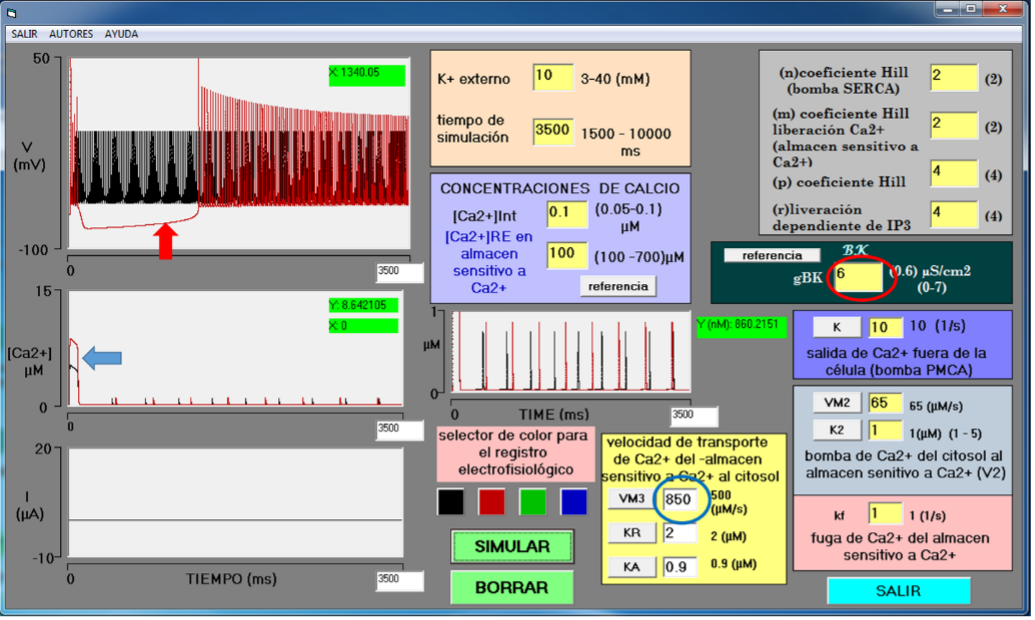
Fuente: Elaboración propia.
Figura 6 Epilepsia simulada por alto K+ externo y ejemplo de pausa del tren de PA por acción del complejo BK/IP3-R. El módulo de entrada muestra un incremento de [K+]e = 10 mM que produce epileptogénesis. El trazo negro en el osciloscopio superior corresponde a una neurona epiléptica por incremento de K+ externo. El trazo rojo en el osciloscopio superior corresponde a la activación del complejo BK/IP3-R por aumento de VM3 de 0 µM/s a 850 µM/s (círculo verde) y de gBK de 0 µS/cm2 a 6 µS/cm2 (círculo rojo). El resultado fue una pausa en el estado epiléptico (flecha roja). Tiempo de simulación 3500 ms.
Bloqueo total de la actividad eléctrica epiléptica en neurona con incremento [K+]e debido al complejo BK/IP3-R
El incremento de la concentración de K+ externo de 3.5 mM a 10 mM genera epileptogénesis. Se simuló el efecto que produce un incremento mayor en la función del complejo BK/IP3-R en este estado epiléptico. Se realizaron experimentos virtuales con un tiempo de simulación de 8 s. Esto permite explorar si en estas condiciones se presenta la despolarización generada por la alta concentración de K+ externo que produce una pausa en el tren de PA, o si la hiperpolarización generada por el complejo BK/IP3-R bloquea completamente el estado epiléptico. En una neurona en condiciones de epilepsia por incremento de la concentración de K+ extracelular (10 mM) y con gBK = 6 µS/cm2 se produce una pausa en el tren de PA con una duración de 543.5 ms cuando VM3 = 830 µM/s y de hasta 6851 ms cuando VM3 = 865 µM/s. Cuando se aumenta VM3 a 870 µM/s se produce un bloqueo total. La hiperpolarización alcanzada por el complejo BK/IP3-R para producir este bloqueo fue de -111.5 mV. Cuando se incrementa gBK a 6.1 µS/cm2 entonces se puede bajar VM3 a 865 µM/s y también se produce un bloqueo total. La hiperpolarización alcanzada fue de -113 mV. Otra condición para bloquear del tren de PA es aumentar gBK a 6.3 µS/cm2 y bajar VM3 a 850 µM/s, la hiperpolarización alcanzada fue de -110 mV. Se observa que existe una interacción entre la cantidad de canales expresada en gBK y la cantidad de Ca2+ que sale del retículo para activarlos y producir un bloqueo total del estado eléctrico epiléptico, siempre que la hiperpolarización sea mayor o igual a -110 mV (en las condiciones del simulador y con un potencial de reposo de -90 mV). A mayor cantidad de canales (gBK) menor VM3 y viceversa. La Figura 7 muestra el caso con VM3 en 850 µM/s y gBK a 6.3 µS/cm2. Se realizaron dos simulaciones: (1) Neurona epiléptica por alto K+ extracelular: en el osciloscopio superior se presenta el registro en esta neurona epiléptica (trazo en negro) por aumento de K+ externo (10 mM) con VM3 = 500 µM/s y gBK = 0 µS/cm2, el resto de las variables permanecen sin cambio; (2) en esta misma neurona se incrementó la actividad del complejo BK/IP3-R. La activación mayor del complejo BK/IP3-R se obtuvo al aumentar gBK de 6 µS/cm2 a 6.3 µS/cm2 (mayor número de canales) y de VM3 de 500 µM/s a 850 µM/s (mayor salida de Ca2+ del retículo). El resto de las variables permanecen sin cambio. En estas condiciones se produce una hiperpolarización que alcanza -110 mV, la cual bloquea totalmente la actividad eléctrica epiléptica (trazo rojo osciloscopio superior). La cantidad de calcio intracelular requerida para detener el estado epiléptico fue de ~ 8.5 µM (flecha azul). En condiciones de reposo, [Ca2+]i oscila entre 50 nM y 100 nM. Incrementos del orden de 19 µM han sido reportados en la cercanía del canal BK en el complejo: canales de Ca2+/canales BK (Cox, 2014).

Fuente: Elaboración propia.
Figura 7 Ejemplo de bloqueo total del estado epiléptico en una neurona por la función aumentada del complejo BK/IP3-R. Se realizaron dos registros: (1) Registro en neurona epiléptica (osciloscopio superior, trazo negro) debido a un incremento de K+ externo (10 mM) con VM3 = 500 µM/s y gBK = 0 µS/cm2 y el resto de las variables sin cambio; se observa un tren de PA sostenidos. (2) Registro en esa misma neurona con activación del complejo BK/IP3-R por un incremento de VM3 de 500 µM/s a 850 µM/s (círculo azul) y de gBK de 6 µS/cm2 a 6.3 µS/cm2 (círculo rojo) con K+ externo de 10 mM y el resto de las variables sin cambio. Se observa un incremento de la concentración de calcio intracelular a 8.5 µM (flecha azul). El resultado fue una hiperpolarización de -110 mV y un bloqueo total el proceso epiléptico (trazo rojo, flecha roja). El eje de voltaje fue llevado a -120 mV. El tiempo de simulación fue de 8 s.
Discusión
La epilepsia es de gran interés clínico. A la luz de los nuevos descubrimientos y nuevas tecnologías se ha propuesto por la Liga Internacional contra la Epilepsia (ILAE, por sus siglas en inglés) una nueva clasificación de las epilepsias con la finalidad de una mejor comprensión de sus aspectos clínicos y contar con una herramienta útil en el ejercicio médico. En dicha clasificación se consideran sus posibles causas con la esperanza de mejorar el diagnóstico y de esa manera implementar un tratamiento específico (Scheffer et al., 2017). Por ejemplo, el diagnóstico de epilepsia por mutaciones de canales iónicos (Tabla 1) ha motivado la investigación con técnicas de biología molecular (Santos et al., 2020). Los hallazgos encontrados incrementaron el estudio de los canales iónicos como blancos terapéuticos (Lerche et al., 2001). Los nuevos resultados han derivado en un gran interés en la investigación experimental con abordajes electrofisiológicos novedosos como los ensambles neuronales (Serrano-Reyes et al., 2020). Se ha puesto énfasis en los mecanismos celulares que generan la epilepsia a dos niveles: (1) redes neuronales, donde se ha encontrado que una fuerte sincronía en la red es una posible causa de la epilepsia (Serrano-Reyes et al., 2020), y (2) mecanismos celulares intrínsecos (Dichter & Ayala, 1987). Sin embargo, poco se sabe sobre los mecanismos celulares que determinan el cese de la actividad eléctrica anómala en las neuronas epilépticas. En la epilepsia focal, cientos de neuronas se sincronizan y presentan una despolarización inusual que lleva a la generación de trenes de potenciales de acción anómalos. Se postula que la hiperpolarización que sigue a este tipo de evento sea responsable de limitar la duración de las descargas interictales epileptiformes (Dichter & Ayala, 1987). En este trabajo se propone que la hiperpolarización generada por la función del complejo BK/IP3-R forma parte de uno de los posibles mecanismos celulares que terminan la duración de la actividad eléctrica epileptiforme en la neurona.
Los resultados sugieren que el complejo BK/IP3-R actúa como un sistema intrínseco de protección en ciertas condiciones anómalas en la neurona. Probablemente se active para tratar de disminuir la excitabilidad en neuronas epilépticas. Cuando el complejo BK/IP3-R no se activa correctamente, el sistema no tendrá ningún efecto protector. Se ha reportado que alteraciones del canal BK, como una disminución en su función, causa epilepsia del lóbulo frontal (N’Gouemo, 2011). En los casos de epilepsia asociada a una disminución en la función de los canales BK, se propone que drogas que activen el canal BK representan una nueva estrategia terapéutica para ciertos tipos de epilepsia (Bentzen et al., 2014). Estos trabajos sugieren la hipótesis de que los canales BK están involucrados en la regulación de la excitabilidad neuronal y control de la aparición de epilepsia. N’Gouemo (2011) formula que los canales BK bloquean la aparición de convulsiones y estados epilépticos. Los resultados del presente trabajo apoyan la propuesta que el complejo BK/IP3-R actúa como mecanismo protector neuronal en ciertos casos (Clements et al., 2013; Pérez-Bonilla et al., 2021). La función del complejo BK/IP3-R y el papel del retículo endoplasmático en la regulación del calcio interno pueden evitar las convulsiones (Karagas & Venkatachalam, 2019). En el presente trabajo solamente se probó el complejo BK/IP3-R en dos mecanismos epileptogénicos: (1) mutación del canal de sodio Nav 1.2, y (2) un incremento de la concentración de potasio extracelular. Los resultados sugieren que la función del complejo BK/IP3-R bloquea completamente o pausa temporalmente la actividad eléctrica epiléptica en la neurona. Sin embargo, el grado de eficacia de este mecanismo en otros tipos de epilepsia podría variar significativamente, sobre todo en casos donde la epilepsia sea generada por redes neuronales con interacciones sinápticas aberrantes entre neuronas (McCormick & Contreras, 2001). Son necesarios estudios experimentales para probar específicamente este mecanismo en diversos modelos animales de epilepsia.
Conclusiones
En el presente trabajo teórico se logró producir epileptogénesis por dos mecanismos: (1) mutación del canal de sodio (NaV 1.2) e (2) incremento de potasio externo. Los resultados obtenidos en las simulaciones con neurona epiléptica a causa de mutaciones en el canal NaV 1.2 muestran que la función del complejo BK/IP3-R está asociada con el bloqueo a la actividad eléctrica epiléptica de la neurona, debido a la hiperpolarización que causa la salida de potasio por los canales BK. Las simulaciones realizadas en neurona epiléptica por incremento de [K+]e muestran una competencia entre la despolarización resultante del aumento de potasio externo y la poshiperpolarización producto de la función de los canales BK. En estas condiciones se produjo una pausa temporal o un bloqueo total de la actividad eléctrica epiléptica de la neurona. Estos resultados confirman un posible mecanismo intrínseco causante del término de la actividad eléctrica epiléptica en la neurona, constituyéndose en un mecanismo protector, como fue sugerido anteriormente (Clements et al., 2013; Pérez-Bonilla et al., 2021). La neurona pone en función toda la maquinaria que regula la homeostasis del calcio interno para incrementar su concentración y activar un gran número de canales BK. En condiciones extremas, se puede incrementar el IP3 y la salida de Ca2+ del retículo, así como disminuir la actividad de las bombas PMCA y SERCA. El grado de efectividad de este mecanismo depende del tipo de epilepsia y puede variar sustancialmente cuando el problema sea un desbalance sináptico en una red neuronal, no abordado en este trabajo. La hipótesis presentada en este trabajo debe verificarse en condiciones experimentales. Este modelo se puede aplicar en estudios con neuronas disociadas a partir de focos epilépticos de procesos neuroquirúrgicos y en otras posibles condiciones experimentales, por ejemplo, en rebanadas de cerebro donde se provoque epilepsia (con drogas convulsivantes o cambios de iones en el espacio extracelular) y se registren las neuronas con cero calcio extracelular y se estimule la salida de calcio del retículo.

