











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La naturaleza hereditaria de todo organismo es definida por su genoma. Debido a la gran importancia de esta molécula y al surgimiento de la tecnología del ácido desoxirribonucleico (DNA, por sus siglas en inglés) recombinante, se han diseñado numerosas pruebas útiles en diversas áreas de investigación, como las ciencias médicas, biológicas, antropológicas, forenses, entre otras, así como en la generación de bancos de DNA y germoplasma que se han establecido con diversos fines (Eguiarte, Souza & Aguirre, 2007).
Entre las técnicas más utilizadas en la manipulación del DNA se encuentra la reacción en cadena de la polimerasa (PCR, por sus siglas en inglés), de la cual existen variantes como la PCR punto final, PCR tiempo real, PCR inversa, PCR anidada, PCR mutagénico, entre otras. Para realizar esta técnica es necesario contar con una muestra libre de proteínas, tanto citoplasmáticas como nucleares que se encuentran asociadas al DNA. Por ello, la extracción del DNA genómico es un paso crucial cuando se desean realizar estudios moleculares, ya que existe variabilidad según el método de extracción utilizado, pues puede proveer contaminación residual con sales y solventes (Blanco-Jarvio, Martínez & Bautista, 2014).
Se han reportado diversas técnicas de extracción con métodos relativamente sencillos y de bajo costo; una de ellas es el método de extracción con fenol-cloroformo-alcohol isoamílico (FCl), considerado uno de los más eficaces para cualquier tipo de muestra. Sin embargo, este procedimiento es laborioso y utiliza compuestos tóxicos que pueden ser peligrosos para el investigador que los manipula, además de contener inhibidores para la reacción de PCR, como el fenol y el cloroformo, por lo que se debe añadir un paso adicional en la extracción, generalmente la filtración en columnas específicas, o realizar varios lavados para eliminar los residuos de los disolventes (Baena, Ramos, Gómez & Gómez, 2013).
Lahiri & Nurnberger (1991) reportaron un método de extracción por gradiente de sales (MgCl2, KCl, NaCl, EDTA, Tris-HCl) para extraer DNA de suficiente calidad para el análisis molecular por Polymerase Chain Reaction-Restriction Fragment Length Polymorphism (PCR-RFLP). Este método reduce la exposición a compuestos químicos peligrosos y, aunque es un método barato y libre de inhibidores, presenta el inconveniente de ser muy laborioso. Daly, Steen, Fairbrother & Idle (1996) reportaron un método que consiste en purificar el DNA mediante la desproteinización con ácido perclórico y la lisis de membranas con un amortiguador rico en sacarosa, MgCl2 y la adición de Tritón X-100 como detergente. Los autores señalan que esta técnica es económica y permite la recuperación de grandes fragmentos de DNA (hasta 30 kb); esta característica es muy importante cuando se requiere amplificar fragmentos de alto peso molecular o generar bibliotecas de DNA genómico.
Por otro lado, se encuentran disponibles a nivel comercial otros productos para purificar el DNA, como es el caso del DNAzol ® o de kits completos, que cuentan con todos los amortiguadores y materiales necesarios para realizar una extracción muy sencilla y en corto tiempo, como los kits de casas comerciales que han desarrollado estos métodos de extracción rápida de DNA; aunque son reproducibles y se obtiene material de alta pureza y peso molecular, la principal limitante del uso de estos métodos es el costo y la poca cantidad de DNA recuperada.
De acuerdo con lo anterior, al elegir el método de extracción del DNA es importante realizar una evaluación detallada del material genómico necesario para la investigación y del procedimiento que se le aplicará a la muestra, ya que los métodos rápidos y automatizados generalmente se utilizan para procesar un número pequeño de muestras que requiere poca cantidad de DNA (ng o pg), mientras que la generación de bancos genómicos y la obtención de material con suficiente calidad para clonación requiere de un reservorio que cuente con una gran cantidad de DNA, a veces en el orden de los µg, que la molécula sea de elevado peso molecular y libre de inhibidores de la PCR (Caboux et al., 2012; Green & Sambrook, 2012).
De modo paralelo, los estudios de epidemiología molecular reportan las frecuencias de ciertos polimorfismos de interés, en poblaciones específicas, que son asociados con la susceptibilidad genética a determinadas enfermedades. Sin embargo, no existen criterios unificados para llevar a cabo la genotipificación, por tanto, la confiabilidad en la asignación de genotipos es variable y depende de la calidad del DNA purificado y de la técnica utilizada para genotipificar, lo que puede conducir a resultados erróneos que tienen bases metodológicas. Se ha establecido que el estándar de oro es la genotipificación por PCR tiempo real (Gaedigk et al., 2015), por su alta sensibilidad y especificidad, no obstante presenta el inconveniente de tener mayor costo, por lo que muchos laboratorios que no cuentan con infraestructura y recursos suficientes recurren a la genotipificación por PCR - RFLP’s, el cual es un método sensible, aunque se debe tener en cuenta la posible presencia de inhibidores de la PCR y de las enzimas de restricción, así como la longitud de los fragmentos amplificados, para tener una mejor resolución del patrón de bandas.
En este contexto, los polimorfismos en los genes del receptor de arilos (AhR), el citocromo 1A1 (CYP1A1) y las glutatión transferasas M1 y T1 (GSTM1 y GSTT1) han sido extensamente estudiados en múltiples poblaciones, y se ha observado una amplia variabilidad en las frecuencias reportadas, conduciendo a resultados inconsistentes con su asociación a cáncer, principalmente (Saitou & Ishida, 2015; Spink et al., 2014). No obstante, es posible que estas inconsistencias sean debidas a errores metodológicos relacionados con la asignación de genotipos, como se mencionó anteriormente, y por lo cual la extracción del DNA y el método de genotipificación presentan alta variabilidad.
En el presente trabajo se compararon los rendimientos de distintas técnicas de extracción de DNA: FCI, GS, GSC, DNAzol ® (cat. DN129) y DNeasy Blood & Tissue Kit ® (cat. 69506), para evaluar los principales parámetros en la calidad del DNA y su posterior uso en el análisis molecular por PCR múltiple, PCR - RFLP’s y PCR tiempo real para analizar la reproducibilidad de las técnicas de genotipificación de los polimorfismos AhR Arg554Lys (rs2066583), CYP1A1 Ile482Val (rs1048943), GSTM1*0 y GSTT1*0 que presentan una amplia variabilidad en la frecuencia, según la población analizada, y para estudiar la asociación a ciertos tipos de cáncer.
MATERIALES Y MÉTODOS
Muestras
Se obtuvieron muestras de sangre periférica de 10 individuos mestizos mexicanos no relacionados, clínicamente sanos. Los participantes firmaron una carta de consentimiento informado; el protocolo fue aprobado por el Comité de Bioética de la Universidad Juárez del Estado de Durango. Las muestras fueron colectadas en vacutainer con EDTA. Se realizaron 5 alícuotas de 1 ml de cada muestra y se mantuvieron en congelación a -20 °C hasta su uso. Cada alícuota de 1 ml se utilizó para extraer el DNA por los distintos métodos. Un total de 50 muestras fueron purificadas por los cinco métodos, es decir, las 10 muestras por cada método, y fueron analizadas por tres distintas técnicas de PCR para genotipificación de polimorfismos.
Métodos de extracción de DNA
Todos los reactivos y solventes utilizados fueron grado biología molecular de la marca Sigma-Aldrich (St. Louis, MO, USA).
Técnica de extracción por Fenol-Cloroformo-Alcohol Isoamílico (FCI) (Green & Sambrook, 2012).
La extracción se llevó a cabo a partir de 1 ml de sangre periférica. Se agregaron 50 μl de SDS al 20%, se mezcló por inversión, se añadió 1 ml de fenol y se incubó 5 min a 65 °C; inmediatamente después se incubó a -20 °C durante 5 min. Se centrifugó la muestra durante 10 min a 10 000 rpm y se recuperó la fase acuosa en otro tubo, donde se agregaron 500 μl de la mezcla FCI en proporción 25:24:1, preparada al momento de adicionarlo. Se centrifugó la muestra a 12 000 rpm y se recuperó la fase acuosa, que fue adicionada a dos volúmenes de etanol absoluto frío. Se centrifugó a 12 000 rpm durante 5 min para recuperar el DNA, después de la evaporación del etanol se adicionaron 50 μl de agua libre de nucleasas y se resuspendió en incubación a 65 °C durante 15 min. Las muestras fueron congeladas a -20 °C hasta su uso.
Técnica de extracción por gradiente de sales (GS) (Lahiri & Nurnberger, 1991)
La extracción se llevó a cabo a partir de 1 ml de sangre periférica, se agregó 1 ml de amortiguador TKM1 (10 mM Tris-HCl pH 8.0, 10 mM KCl, 10 mM MgCl2, 2 mM EDTA) y 25 μl de Igepal. Se agitó vigorosamente y se centrifugó a 2200 rpm durante 10 min. Se desechó el sobrenadante, la pastilla se lavó con 1 ml de amortiguador TKM1 y se centrifugó a 2200 rpm durante 10 min. Se realizaron los lavados hasta obtener la fracción blanca. Se desechó el sobrenadante y la pastilla se resuspendió en 160 μl de amortiguador TKM2 (10 mM Tris-HCl pH 8.0, 10 mM KCl, 10 mM MgCl2, 2 mM EDTA, 0.4 M NaCl). Se adicionaron 10 μl de SDS 10% y se incubó a 55 °C durante 10 min. Posteriormente se agregaron 120 μl de NaCl 3 M y se centrifugó a 12 000 rpm durante 5 min. Se recuperaron 500 μl del sobrenadante y se adicionaron a otro tubo que contenía dos volúmenes de etanol absoluto. Se mezcló por inversión varias veces hasta observar la hebra de DNA precipitada por el etanol y se recuperaron las hebras, que fueron lavadas con etanol frío al 70%. Se recuperó el DNA y después de la evaporización del etanol se adicionaron 50 μl de agua libre de nucleasas. Se resuspendió en incubación a 65 °C durante 15 min. Las muestras fueron congeladas a -20 °C hasta su uso.
Técnica de extracción por gradiente de sacarosa (GSC) (Daly et al., 1996)
La extracción se llevó a cabo a partir de 1 ml de sangre periférica a la que se le agregaron 9 ml de amortiguador de lisis (320 mM sacarosa, 5 mM MgCl2, 1% Tritón X-100, 10 mM Tris - HCl pH 7.4), se agitó vigorosamente y se centrifugó a 2000 rpm por 5 min. Se decantó el sobrenadante y se agregaron 400 μl de amortiguador de suspensión (150 mM NaCl, 60 mM EDTA, 1% SDS, 400 mM Tris - HCl pH 7.4) con 100 μl de NaClO4 5 M. La suspensión fue mezclada en un agitador rotatorio por 15 min a temperatura ambiente (TA) e incubada a 65 °C por 30 min. Posteriormente, se le agregaron 400 μl de cloroformo a -20 °C y la mezcla se mantuvo en agitación por 10 min, seguido de centrifugación a 1400 rpm por 10 min. Se adicionaron dos volúmenes de etanol absoluto a la fase acuosa y se mezcló hasta que la hebra de DNA precipitara. Se recuperaron las hebras y se lavaron con etanol al 70%. Se adicionaron 50 μl de agua libre de nucleasas y se resuspendió en incubación a 65 °C durante 15 min. Las muestras fueron congeladas a -20 °C hasta su uso.
Extracción con DNAzol ® BD de Molecular Research Center (Cat. DN129)
Se siguieron las especificaciones del fabricante, las cuales se detallan a continuación. La extracción se llevó a cabo a partir de 500 μl de sangre periférica, se le agregó 1 ml del reactivo DNAzol. Se mezcló por 20 s y se incubó a temperatura ambiente (TA) por 5 min. Se agregaron 400 μl de isopropanol, se agitó en vórtex y se incubó 5 min a TA. Se centrifugó la muestra a 8000 rpm durante 6 min. Se decantó el sobrenadante y se reservó la pastilla, se agregaron 500 μl de DNAzol y se mezcló hasta que se incorporara con la solución. Se centrifugó la muestra a 8000 rpm durante 5 min, se decantó el sobrenadante y se reservó la pastilla. Se agregó 1 ml de etanol al 75% y se centrifugó a 8000 rpm durante 5 min. Se decantó el sobrenadante y se adicionaron 50 μl de agua libre de nucleasas, el DNA fue resuspendido en incubación a 65 °C durante 15 min. Las muestras fueron congeladas a -20 °C hasta su uso.
Extracción con DNeasy Blood & Tissue de Quiagen ® (Cat. 69506)
Se siguieron las especificaciones del fabricante, las cuales se detallan a continuación. La extracción se llevó a cabo a partir de 200 μl de sangre periférica, se agregaron 20 μl de proteinasa K y se mezclaron por inversión. Se agregaron 200 μl de amortiguador AL y se agitó en vórtex. Se incubaron las muestras a 56 °C durante 10 min. Se agregaron 200 μl de etanol absoluto a TA y se agitó en vórtex. En un tubo nuevo se colocó una columna con filtro DNeasy y se adicionó la muestra colocándola en el centro del filtro. Se centrifugó a 8000 rpm durante 1 min. Se desechó el sobrenadante y se pasó la columna a un tubo nuevo, agregándole 500 μl de amortiguador AW1, se centrifugó a 8000 rpm durante 1 min, se desechó el sobrenadante y se conservó la columna. Se realizó el procedimiento anterior adicionando 500 μl de amortiguador AW2. Se centrifugó por 3 min a 14 000 rpm, se desechó el filtrado y finalmente se pasó la columna a otro tubo nuevo para adicionar 200 μl de amortiguador AE. Se incubó durante 1 min a TA para permitir que el DNA se desprendiera de la columna y pudiera ser eluído en la siguiente centrifugación que se realizó a 8000 rpm durante 1 min. Las muestras fueron congeladas a -20 °C hasta su uso.
Cuantificación de DNA, análisis de integridad y pureza
Se analizó la cantidad y pureza del DNA por nano-espectrofotometría en un nanodrop 2000 de Thermo Fisher ®. Los valores reportados como óptimos en la pureza del DNA son entre 1.7 - 1.9 (Green & Sambrook, 2012).
La integridad fue analizada por electroforesis en gel de agarosa al 1% teñido con 10 μg/ml de bromuro de etidio, y fue visualizado en un fotodocumentador MiniBis Pro ® (Accesolab), bajo luz ultravioleta (UV). Se utilizó el marcador de peso molecular 1 Kb de AXYGEN ®.
Genotipificación de polimorfismos
Para el análisis de PCR múltiple se amplificaron tres genes polimórficos simultáneamente; los oligonucleótidos Fwd gaactccctgaaaagctaaagc, Rvs gttgggctcaaatatacggtgg que amplifican un fragmento de 480 pb que corresponde al gen GSTT1; como control interno se utilizaron los oligonucleótidos Fwd gaactgccacttcagctgtct, Rvs cagctgcatttggaagtgctc que amplifican 312 pb del gen CYP1A1; y finalmente los oligonucleótidos Fwd ttccttactggtcctcacatctc, Rvs tcaccggatcatggccagca que amplifican un fragmento de 215 pb del gen GSTM1. Los oligonucleótidos fueron diseñados en la región polimórfica de los genes GSTM1 y GSTT1, donde ocurre una deleción; los individuos que tienen el polimorfismo en condición homóciga resultan negativos para la reacción de PCR, sin embargo, los heterócigos y los homócigos silvestres amplifican el mismo fragmento resultando positivos para la reacción (Abdel-Rahman, el-Zein, Anwar & Au, 1996).
Para el análisis de PCR - RFLP se utilizaron los oligonucleótidos Fwd ctgtctccctctggttacaggaagc y Rvs ttccagccgttgcagcaggatagcc que amplifican un fragmento de 204 pb del gen CYP1A1. El polimorfismo CYP1A1 Ile482Val (rs1048943) fue identificado por la longitud de los fragmentos de restricción utilizando 0.4 unidades de la enzima BsrDI, donde el homócigo silvestre genera fragmentos de 149 pb y 55 pb, el heterócigo 204 pb, 149 pb y 55 pb, mientras que el homócigo polimórfico pierde el sitio de restricción y se observa una banda de 204 pb (Cascorbi, Brockmöller & Roots, 1996). La visualización de los fragmentos se realizó después de la electroforesis en gel de agarosa al 3%, posterior tinción con bromuro de etidio 10 μg/ml. Se utilizó el marcador de peso molecular de 50 pb de Invitrogen ®.
En el PCR tiempo real se genotipificó el polimorfismo AhR Arg554Lys (rs2066583) del receptor de arilos, utilizando la sonda TaqMan C_11170747_20X de Applied Biosystems ® siguiendo las recomendaciones del fabricante, en un PCR tiempo real Step One de Applied Biosystems ®, la asignación de genotipos la realiza el equipo de forma automática según los fluoróforos detectados durante la reacción.
Secuenciación e identidad de fragmentos amplificados
Se utilizaron los oligonucleótidos Fwd ggcaagataatactgcaccga, Rvs agcttgagttcagagccaagg para amplificar un fragmento de 345 pb del gen AhR y los oligonucleótidos antes descritos para obtener los fragmentos de 480 pb, 215 pb y 204 pb correspondientes a los genes GSTT1, GSTM1 y CYP1A1, respectivamente, que fueron reamplificados y purificados utilizando el Kit Purification Product of PCR ® de Qiagen para su secuenciación, la cual se llevó a cabo en la Unidad de Secuenciación del Instituto de Investigaciones Biomédicas de la Universidad Nacional Autónoma de México (UNAM). Para verificar la identidad de las secuencias se realizó un análisis nucleotide blast con el software blastn disponible en la página del National Center for Biotechnology Information (2015).
Análisis estadístico
Se analizaron las medidas de tendencia central (Media ± DE) para los parámetros de pureza y cantidad, posteriormente se aplicó la prueba de Shapiro-Wilks para verificar la normalidad de la distribución y una prueba no paramétrica de Kruskal-Wallis usando el paquete estadístico SPSS V-20.0 para Windows.
RESULTADOS
Los parámetros más importantes que se deben tomar en cuenta en la extracción del DNA son la pureza, cantidad e integridad. En la pureza, los métodos GS, DNAzol ®, DNeasy ®, GSC y FCI tuvieron valores de 1.71 ± 0.11; 2.52 ± 0.70; 1.77 ± 0.08; 1.80 ± 0.04 y 1.74 ± 0.24, respectivamente; mientras que en la cantidad expresada en ng/μl se obtuvieron 136.76 ± 115.18; 29 ± 28.8; 23.19 ± 7.42; 211.93 ± 103.93 y 189.02 ± 57.87 para GS, DNAzol ®, DNeasy ®, GSC y FCI, respectivamente (tabla 1). En la comparación entre grupos no se encontraron diferencias significativas en la cantidad de DNA obtenida por μl (p = 0.215), mientras que en la pureza se encontró diferencia en el método DNAzol ® con respecto a los demás (p = 0.05).
Tabla 1 Parámetros de pureza y cantidad de DNA obtenidos en distintos métodos de extracción.

Fuente: elaboración propia.
Adicionalmente, se analizó la cantidad total de DNA extraído y se observó alta variabilidad debido a que los métodos tradicionales recuperan mayor cantidad, hasta 83 μg en el caso de GS, con respecto a los kits comerciales que recuperan alrededor de 6 μg (p = 0.03) (figura 1).

Fuente: Elaboración propia.
Figura 1 El método GS recupera 54.6 µg ± 46.0, DNAzol 8.7 µg + 8.6, DNeasy 4.6 µg ± 1.48, GSC 21.1 µg ± 10.3 y FCI 9.4 µg ± 2.8 (p = 0.03).
La integridad del DNA se analizó por electroforesis y se observó DNA de elevado peso molecular en todas las muestras aisladas por los diferentes métodos (figura 2).

Fuente: Elaboración propia.
Figura 2 Gel de integridad de DNA. Se muestran cinco muestras representativas, purificadas por los diferentes métodos. Para visualizar bien las bandas se cargaron ~200-250 ng de DNA por carril, de las muestras de GS, FCI y GSC se cargaron de 1-2 µL, mientras que de DNAzol se cargaron 10-20 µL y de DNeasy de 10-15 µL.
Para el análisis molecular se seleccionaron cuatro genes que se encuentran ampliamente reportados en distintas poblaciones y cuya genotipificación se ha realizado por diversas metodologías. En el análisis molecular por PCR y la asignación de genotipos se incluyeron todas las muestras purificadas por los distintos métodos de extracción de DNA mencionados, de tal modo que la misma muestra fue purificada por cinco métodos distintos y fue genotipificada para los genes AhR, CYP1A1, GSTM1 y GSTT1 por tres metodologías estándar (figura 3).

Fuente: Elaboración propia.
Figura 3 Métodos de genotipificación por PCR. A) PCR múltiple para las deleciones en los genes GSTM1 y GSTT1. a) Las muestras de los carriles 5, 6, 7 9 y 11 son positivos para ambos genes, las muestras de los carriles 2 y 3 son positivas para GSTT1, la muestra del carril 4 es negativa para ambos genes y las muestras de los carriles 8 y 10 no amplificaron, tampoco amplificó el control interno. b) PCR - RFLP’s del polimorfismo CYP1A1 Ile462Val. Las muestras de los carriles 1, 2, 3 y 6 son heterócigas con genotipo Ile/Val, las muestras del carril 7 y 8 son homócigas Val/Val y la muestra del carril 9 es homóciga Ile/Ile, el control negativo se observa en el carril 4. c) PCR tiempo real para la genotipificación del polimorfismo AhR Arg554Lys. Las muestras en el eje X corresponden a homócigos Lys/Lys, las muestras en el eje Y corresponden a homócigos Arg/Arg, mientras que los intermedios corresponden a los heterócigos Arg/Lys, la asignación de genotipos es automática, según el fluoróforo detectado durante la reacción.
Para validar los resultados anteriores, se secuenciaron las muestras homocigotas para ámbos alelos de cada polimorfismo; en el caso de GSTM1 y GSTT1 se secuenciaron muestras positivas para ambos genes y se realizó un análisis nblast que confirmó la identidad de las secuencias. En la figura 4 se observan los cambios de un solo nucleótido en el gen AhR Arg554Lys (rs2066583) y CYP1A1 Ile482Val (rs1048943).

Fuente: Elaboración propia.
Figura 4 Secuencias del gen AhR Arg554Lys. a) Triplete que codifica para el aminoácido lisina (AAA) y b) Triplete que codifica para el aminoácido arginina (AGA). Secuencias del gen CYP1A1 Ile482Val. c) Triplete que codifica para el alelo valina (GTT) y d) Triplete que codifica para el alelo isoleucina ATT).
En la genotipificación de GSTM1*0 y GSTT1*0 por PCR múltiple se observó que las muestras purificadas por el método de GS y GSC tuvieron una eficiencia de amplificación del 100% y una concordancia también del 100%; es decir, el resultado fue reproducible y la muestra presentó el mismo genotipo, independientemente del método de extracción. En el caso de las muestras purificadas por DNAzol ® y DNeasy ® se observó una eficiencia de amplificación del 80% y una concordancia del 100%, mientras que las muestras purificadas por el método FCI no amplificaron, incluso después de tres intentos (tabla 2).
Tabla 2 Porcentaje de la eficiencia de amplificación y concordancia entre genotipos de GSTM1*0, GSTT1*0, CYP1A1 Ile462Val y AhR Arg554Lys.
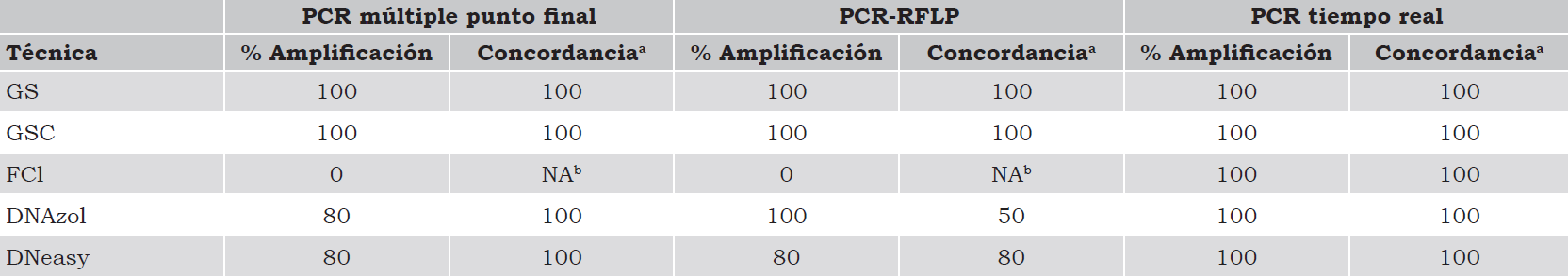
a Se consideró concordancia cuando el resultado fue reproducible y la muestra presentó el mismo genotipo, independientemente del método de extracción; b NA: no aplica.
Fuente: Elaboración propia.
En la genotipificación de CYP1A1 Ile462Val por PCR-RFLP’s se obtuvo una eficiencia de amplificación del 100% en los métodos GS, GSC, del 80% para DNAzol y DNeasy y de 0% en el método FCI. Sin embargo, la concordancia fue del 100% entre los métodos GS y GSC, del 80% en el método DNeasy ® y del 50% con DNAzol ®.
Finalmente, en la genotipificación de AhR Arg554Lys por PCR tiempo real se obtuvo una eficiencia de amplificación del 100% en todas las muestras y una concordancia del 100% entre los distintos métodos (tabla 2).
DISCUSIÓN
La calidad del DNA purificado es un punto clave en la aplicación de diversas técnicas moleculares. Los principales parámetros analizados después de una extracción son la pureza, la cantidad y la integridad. Se evaluaron tres métodos tradicionales de extracción de DNA y dos métodos comerciales, así como su eficiencia de amplificación y concordancia para genotipificar polimorfismos en los genes GSTM1, GSTT1, CYP1A1 y AhR, que son de gran interés toxicológico.
Una técnica de extracción ideal es aquella que posee un número limitado de pasos, mínima utilización de disolventes peligrosos, requerimiento limitado de equipos y que sea relativamente económica (Osorio-Cadavid, Ramírez, López & Mambuscay, 2009). La técnica de FCl requiere fenol que es un disolvente tóxico e irritante, además puede causar quemaduras graves en la piel y serias lesiones oculares. Adicionalmente, el cloroformo ha sido descrito como potencial carcinógeno humano capaz de causar la muerte si no es usado de manera adecuada; todas estas características pueden afectar la salud de la persona que manipula las muestras durante el proceso de extracción.
En contraste, las técnicas de extracción por sales y sacarosa utilizan reactivos inocuos como sacarosa, MgCl2, NaCl, EDTA y Tris-HCl; aunque no se han reportado casos de toxicidad grave, el SDS es clasificado como un compuesto moderadamente tóxico en concentraciones superiores a 1000 mg/Kg. En el método GS se requiere este reactivo a una concentración del 10%, que no representa un riesgo grave a la salud. Por su parte, el NaClO4 es empleado en el método gradiente de sacarosa a una concentración de 5 M; este reactivo es clasificado como una sal oxidante fuerte que puede provocar incendios o explosiones al no ser utilizado adecuadamente; mientras que los kits comerciales son seguros y reproducibles, aunque en el caso de DNAzol ® no se obtuvieron buenos resultados; no obstante, estos reactivos tienen costos elevados, lo que podrían ser una limitante en la ejecución de un proyecto que involucra el análisis de un alto número de muestras.
La pureza del DNA extraído se encontró dentro de los rangos establecidos como óptimos (1.7 - 1.9), a excepción de las muestras purificadas con DNAzol ® que mostraron valores de hasta 2.5. Estos valores no se consideran confiables, pues en este caso puede haber contaminación con proteínas y/o disolventes que pudieran interferir en la reacción de PCR, como se ha documentado (Green & Sambrook, 2012).
En la cantidad de DNA extraído se obtuvieron concentraciones desde 8.5 ng/μl de DNA en los kits comerciales hasta 393.20 ng/μl en el método GS, sin embargo, las diferencias no fueron significativas debido a la alta variabilidad entre los métodos; aunque en el caso de DNeasy ® se observaron desviaciones muy pequeñas, como es esperado en un método comercial que debe ser reproducible.
En cuanto a la cantidad total, Caboux et al. (2012) reportaron que, utilizando la técnica de extracción adecuada, es posible obtener hasta 300% más material genético, alcanzando un rendimiento de hasta 45 μg de DNA total, partiendo de 1 ml de muestra sanguínea. En este estudio, el método que recuperó mayor cantidad de DNA fue el método GS (μ = 54.6 ± 46.0 μg). A este respecto, en algunas investigaciones es muy importante obtener la mayor cantidad de muestra, por ejemplo, para la creación de bancos genómicos es necesario resguardar una cantidad adecuada de material y una mínima contaminación con proteínas y sales o disolventes. Por otro lado, los aductos en el DNA son un marcador de efecto ante la exposición a diversos tóxicos, para su determinación se requieren cantidades de DNA en el orden de los μg, por lo que la extracción debe ser muy eficiente (Monien et al., 2014); en estos casos las técnicas de extracción por GS y GSC ofrecen esta ventaja.
Por otro lado, los estudios de epidemiología molecular y farmacogenómica analizan la frecuencia de ciertos genotipos en diversas poblaciones, con la finalidad de establecer asociaciones entre la exposición a xenobióticos, la capacidad metabólica, la presencia de biomarcadores de efecto y el desarrollo de ciertas patologías, sin embargo, los resultados son inconsistentes en la mayoría de los casos; la falta de reproducibilidad puede ser atribuida al método de extracción de DNA y/o a la técnica utilizada para la genotipificación. A este respecto, el proyecto internacional HapMap ha sugerido estandarizar las técnicas para que los resultados puedan ser comparables y confiables, de tal modo que podría haber consistencia de resultados entre poblaciones, ya que actualmente estas diferencias pueden ser debidas a errores metodológicos (Garte, 1998; International HapMap Project, 2015).
Los genes AhR, CYP1A1, GSTM1 y GSTT1 han sido genotipificados en poblaciones caucásicas, asiáticas, africanas y en poblaciones con diversos grados de mestizaje (Dong et al., 2008; He et al., 2014; Pérez-Morales et al., 2014). Las frecuencias reportadas son muy variables y los estudios de asociación, principalmente con cáncer, son inconsistentes. No obstante, los estudios realizados en países desarrollados se llevan a cabo por análisis de fusión a alta resolución (high resolution melting analysis [HRM]), discriminación alélica con sondas TaqMan ®, PCR alelo específico (AS-PCR) y pirosecuenciación (Gaedigk et al., 2015; Zhang, Zhao, Cui, Wang & Wang, 2015); estos métodos tienen alta reproducibilidad en cualquier tipo de muestra de DNA, aunque son muy costosos y requieren infraestructura de vanguardia, por lo que en los países poco desarrollados el acceso a esta tecnología está limitada, siendo necesario analizar los resultados obtenidos con las técnicas tradicionales, menos costosas y compararlas con el estándar de oro.
En la genotipificación de GSTM1*0 y GSTT1*0 por PCR múltiple se observó una concordancia del 100% entre los resultados obtenidos por los métodos GS, GSC, DNAzol ® y DNeasy ®, mientras que las muestras purificadas por el método FCI no amplificaron, posiblemente debido a que el fenol o el cloroformo inhiben la PCR, como se ha reportado previamente por otros autores (Thompson, Duncan & McCord, 2014).
En la genotipificación de CYP1A1 Ile462Val por PCR - PFLP’s se obtuvo una concordancia del 100% entre los métodos GS y GSC, del 80% en el método DNeasy ® y del 50% con DNAzol ®; es posible que la baja cantidad de DNA obtenida mediante DNeasy ® pueda producir un bajo número de copias del amplicón y esto interfiera con el análisis de restricción; en el caso de DNAzol ® la eficiencia de amplificación fue del 100% y la concordancia del 50%, debido posiblemente a que la contaminación residual, reflejada en los valores 260/280 nm, no actúe como inhibidor de la polimerasa, pero sí como inhibidor de las enzimas de restricción, ya que se observaron digestiones parciales.
Finalmente, en la genotipificación de AhR Arg554Lys por PCR tiempo real se obtuvo una eficiencia de amplificación del 100% en todas las muestras y una concordancia del 100% entre métodos, lo que demuestra que esta metodología es muy sensible, aun en muestras que no son amplificadas por el PCR punto final (debido a la presencia de posibles contaminaciones); además de que utiliza cantidades de DNA muy pequeñas, incluso de 5 ng - 50 ng por reacción. Los estudios reportados por Zhang et al. (2015) y Gaedigk et al. (2015) comprobaron que la genotipificación con sondas TaqMan ® es muy sensible y confiable, incluso en locus altamente polimórficos como el CYP2D6, además de ser tan eficiente como otras metodologías más sofisticadas, entre ellas la pirosecuenciación, HRM y AS-PCR, con la ventaja de tener un costo más bajo que las técnicas antes mencionadas.
Los resultados obtenidos en este trabajo demuestran la importancia de elegir un método de extracción de DNA adecuado y considerar su impacto en la tipificación de genotipos, ya que las bases metodológicas de estas técnicas son extendidas al diagnóstico molecular, y al no ser considerados estos factores los resultados pueden resultar erróneos.
CONCLUSIONES
Los métodos de extracción por GS y GSC tuvieron los mejores resultados en pureza y cantidad. Las muestras amplificaron en todas las variantes de PCR. La genotipificación por medio de sondas TaqMan ® mostró una eficiencia de amplificación y genotipificación del 100%, independientemente del método de extracción.

