











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkModern cardiovascular medicine is a magnificent set of advanced scientific knowledge, technological marvels, old and modern accurate diagnostic procedures, proven therapeutic interventions and a bouquet of intuitive but rational ideas and concepts. Medicine, clearly, is neither a religion nor a closed and unmodifiable doctrinal body. On the contrary, as a liberal discipline with a marked vocation to become more and more a rigorous applied science; the medicine of our time is widely opened to constant reviews and renewals of the paradigms that are the foundations of our daily work. However, the scientific method and its derivative, the evidence-based medicine must preside over any attempt to change or modify current paradigms or principles that have proven to be truthful and useful during decades. In some industrialized and affluent nations as the United States and some European countries, we have observed, from time to time, the recurrent obsession to propose radical changes in some practices or concepts profoundly rooted in medical imaginary and clinical knowledge. These attempts to behead long-time honored paradigms can be properly called an iconoclastic attitude. Iconoclast, according with The Merriam-Webster Dictionary, is «a person who destroys religious images or opposes their veneration, or someone who attacks settled beliefs or institutions».1 Although there is nothing sacred or immovable in science, if someone pretends, in a preposterous and absurd example, overthrow the established fact that the earth is spherical, arguing that it seems to be flat as far as the eye can see, the revisionist has the entire burden of proof and has to demonstrate, beyond any rational doubt, the truthiness of that assertion with unchallengeable evidences. Certainly, medical science has no sacrosanct beliefs or dogmas, but holds proven facts; it does not worship ideas, intellectual leaders or institutions, but it supports demonstrable and measurable principles, i.e., the pristine scientific truth, although we know that as all truths, the scientific one is always relative and partial.
In this context, there is nowadays an insistent campaign against the cholesterol hypothesis, which tries to deny the concept that binds the rise of serum LDL cholesterol concentrations with the development of atherosclerotic lesions. However, the iconoclasts in their attempt to overthrow the established atherogenic concepts, must not only destroy the previous paradigms, but have to build new ones. They have not only to denounce the inconsistencies and flaws they seem to find in the cholesterol hypothesis, but instead they have to construct their own massive building of animal experiments, epidemiological evidences and clinical and therapeutic experiences, showing all them up that cholesterol is not involved in the origin and growth of atherosclerotic plaques, proposing as a consequence, new working hypothesis of atherogenesis. This is a challenge that revisionists, of course, will never be able to accept or fulfill. But, alas, as it is known, it has always been easier to destroy than to build up. Notwithstanding, in recent times, a handful of studies, opinions and meta-analyses have been published, aimed to shake (and eventually dethrone) the basic concept on which primary and secondary atherosclerosis prevention is based. The debate has exceeded the strictly scientific limits, becoming sometimes insulting or slandering, and by the hand of certain sensationalist media and net blogs has affected broader audiences, sowing doubts and fears in many lay persons and causing sometimes the abandonment of treatments or the annoying disagreement among patients and their physicians. Also, in their attempt of take down the cholesterol role in atherosclerosis, the revisionists try to replace the lipid mechanistic atherogenic hypothesis with the pathophysiological role of other well-known contributing factors as stress, hyperglycemia, and inflammation, raising the false dilemma of choosing between cholesterol and other risk factors, as causative agents of atherogenesis. Atherosclerosis is indeed a biopathological drama played by a large cast composed by main and supporting actors. None of these factors or conditions is mutually exclusive with hypercholesterolemia, but indeed all of them are complementary or accessory contributing elements in the atherosclerotic plaques building up. And precisely this notion, recognized long time ago, gives foundation to the concept of holistic prevention, whose battle´s cry could be: put down, at the same time, all cardiovascular risk factors not leaving a single one stand.
This reflection paper just want to point out and criticize several of the iconoclastic revisionists’ claims appeared in the past and recently in some reputable medical journals. Also, this work complements the position of some of the societies agglutinated under this Journal (ANCAM, AMPAC, ANCISSSTE, ANCCMR), recently published.2
There are three outstanding areas subjects to revisionist criticism: 1) The cholesterol hypothesis. 2) The role and safety of statins, ezetimibe, and pro-protein convertase subtilisin-kexin type 9 (PCSK9) inhibitors, in primary and secondary coronary prevention, and 3) The role of dietary cholesterol and saturated fat in the genesis of atherosclerosis.
In the first section of this clinical reflection we will review the conflicting points of the cholesterol hypothesis, refuting the arguments of the revisionists. As a matter of fair play, it is time now for us to look over the thesis of the iconoclasts.
1. The cholesterol hypothesis
The following are some of the more conspicuous revisionist arguments against the cholesterol hypothesis (Table 1):
Table 1: Some iconoclastic arguments against the cholesterol hypothesis.
| 1. Experimental studies, linking atherosclerosis with high cholesterol intake, only has given positive results in herbivores, not in omnivores or carnivores, like human beings |
| 2. The seminal study directed by Ancel Keys (the Seven Countries Study, 1958) must be rejected as it is considered to be flawed by today’s standards. So, the relation among diet cholesterol-saturated fat intake and coronary heart disease lacks all sustenance |
| 3. Goldstein and Brown prophecy (statins will end coronary atherosclerotic disease as a relevant public health problem) has not been fulfilled |
| 4. Lowering cholesterol levels does not improve mortality rates |
| 5. Lessening cholesterol concentrations in the elderly is not useful. High cholesterol serum concentrations help survival |
An iconoclast argument: cholesterol does not cause coronary heart disease.3 The reluctance to accept the cholesterol hypothesis is nothing new. The amazing discoveries of legendary Russian scientist Nikolai N. Anichkov, who in 1913, via elegant experiments showed the role of cholesterol in the genesis of atherosclerosis,4 has been rampantly negated or belittled even to these days, partly for ignorance but also for racists and political prejudices. This contemptuous attitude delayed more than half a century the setup of lipid lowering strategies for coronary prevention, fact that indirectly was the cause of hundreds of thousands of foreseeable and untimely deaths. Still in the 70s, someone could claim, as Olivier did in a famous British medical journal, that «the view that raised plasma cholesterol is per se a cause of coronary heart disease is untenable».5 But in the present times, is hardly conceivable that despite the colossal mountain of evidence of all kinds, there are still researchers and clinicians who had the nerve to deny the validity of the cholesterol hypothesis.
Experimental studies do not support the cholesterol hypothesis, except for vegetarian animals. Those who claim that atherosclerosis is not observed in carnivores, naturally or experimentally, are hoisting an apparently obvious remark, which is however not entirely truthful. To begin with, human beings are not natural carnivores or omnivores. On the contrary, we are genetically herbivorous and frugivorous animals, sharing more than 95% of our genome with chimpanzees and bonobos, our closest relatives, also fundamentally frugivorous, although occasionally they could hunt smaller animals and devour their meat. At the dawn of the irruption of hominids in the planet, our remote ancestors were fundamentally vegetarians. Probably, the inclusion of meat in the usual diet started since the rise of the most direct human antecessors, the Australopithecus gender and the ancient archaic men (Homo habilis), who began to ingest the carrion of the victims of true predators. But later on, humans evolved from being simply scavengers to becoming the most ruthless and effective hunters in the natural world, thanks to the weapons they could built with their skillful hands (with the thumb opposed to the rest of the fingers), their intelligence, the use of language as a communication tool, and social coordinated cooperation. For long millennia, our more recent predecessors developed worldwide, a tribal culture based in hunting and gathering activities, with no cereals as frequent foods. The noticeable success of that form of wandering living came to an end, when flourishing human communities grew to the point that they exhausted both the game reserve and the natural provision of plant foods. This drastic change in the mode of food producing was also forced by the adverse cyclic climatic changes that have always affected the planet. However the way it was, human ingenuity solved that situation with the invention of agriculture and domestication of livestock, approximately 10,000 or 20,000 years ago, yielding to the worldwide emergence of civilized and urban life, and the notion of the nation-states: the era of the splendorous classical cultures, characterized, among many other traits, by the introduction of an omnivorous diet, the phenotypic modification of the livestock, the introduction of cereals as the base of nutrition, the use of salt in cooking and food preserving, as well as the manufacture and consumption of beer, wine, and other alcoholic beverages.6 The contradictions between our genetic inheritance (as forage-eater beings) and the new social and cultural style of life and nourishment, explain the surge of the so-called «civilization diseases»7 (hypertension, atherosclerosis, obesity, diabetes mellitus type 2, and the like), that are, nowadays, some of the most important challenges of public health around the world.6,7 Then, the truth is that we are genetically vegetarian animals, transformed into omnivores for social and historical reasons, and we have adopted an omnivorous diet only in the last thousands of years of our very long stay on the planet.
On the other hand, although atherosclerosis is practically inexistent in reptiles and other cold-blood terrestrial animals, does develop indeed in a wide spectrum of other species and genders.8 For example, through several experimental models it is possible to cause atherosclerotic lesions in herbivorous mammalians as rabbits and also in animals which are basically herbivorous, but which occasionally or frequently can be converted in omnivorous as anthropoid monkeys, mice, rats, and Guinea pigs.9 Also, atherosclerosis can develop spontaneously in grain-eating birds, like hens, turkeys and pigeons, and also in carnivores birds of prey.8-10 To contradict the revisionist’s ideas, atherosclerosis develops also in natural carnivores like dogs and cats.8-12 Experimental atherosclerosis is achieved using high cholesterol diets, blocking thyroid function, the administration of other hormones, hypertension, stress, or genetic manipulation, among many other interventions.9-11 Also, atherosclerotic lesions could be seen spontaneously in some big sea mammalians as cetaceous and even in carnivorous marine animals as some pinnipeds (sea lions and seals).13 Therewithal, in some fishes as salmons, atherosclerotic lesions can be found, sometimes spontaneously and also experimentally by the induction of a diet slightly rich in fat.14 So, it is not true that atherosclerosis only affects herbivorous; on the contrary, it can affect a wide range of other animals (including fish-eating species and strict carnivores), either in natural form or experimentally induced.
Particularly important in this context is the strong evidence, fully supporting the hypothesis of cholesterol, given by the Watanabe heritable hyperlipidemic rabbit, with a mutant modification of the LDL receptor and severe hyperlipidemia.15 Mainly the rabbits of the strain known as the myocardial infarction prone-Watanabe heritable hyperlipidemic animals (WHHLMI), even with their normal strictly herbivorous diet, spontaneously develop initially fatty streaks, but later on, full-blown vulnerable fibro-atheromatous lesions, complicated by myocardial infarctions.16,17 In many models of experimental atherosclerosis, diverse transgenic/knockout animals have been used, which develop atherosclerosis faster and more severe than their wild-type counterparts, inclusively without an atherogenic diet.18 The modern ADN techniques permit to generate these types of experimental animals, mimicking certain dyslipidemias seen in humans.19 But in both, the genetically altered animals and their wild types counterparts, the addition of important amount of cholesterol in the chow, causes and accelerates the atherogenic process.20 What more additional comparative physiologic and experimental data are needed to further verify the cholesterol hypothesis?
The pathological evidence about the cholesterol involvement in atherogenesis is also overwhelming. As Daniel Steinberg has stated in his book on the «wars of cholesterol»,21 since the beginning of this saga there was enlightened pathological evidence of the embroilment of cholesterol and inflammatory cells in the development of atherosclerotic lesions. Anichkov, in fact, described the whole spectrum from monocytes to fat-laden foam cells in arterial lesions, whose formation is one of the first phenomena in the atherogenesis pathway. The monocyte/macrophage origin of foam cells was proven many years later with the development of sophisticated monoclonal antibodies techniques.22 Nowadays, it is known that not monocytes, but macrophages have scavenger receptors that allow these cells to retire extracellular oxidized sub-endothelial LDL-cholesterol, leading them to the intracellular storage of fat, phenomenon characteristic of atherosis, the typical lesion of the fat streak, and a primeval step to atherosclerosis.23-26 Thanks to the seminal contributions of Vogel, Ignatowski, Virchow, Rokitansky, Anichkov and Windaus, among others, since many years ago, we have accumulated solid evidence about the cholesterol involvement and inflammation in the genesis of the atheromatous process, as well as the importance of vascular calcification and the role of thrombosis in the progression of the lessions.27 In recent times, the paramount works of Ross and Libby, along with a pleiade of illustrious researchers, have firmly established the foundations of our thorough understanding of the complex atherogenic process.28-31 The most compelling evidence points out that when the concentrations of lipoproteins rich in cholesterol and ApoB 100, are elevated it is stimulated the traffic of lipoproteins to the sub-endothelium, where they are trapped and modified by oxidation. This original phenomenon, the deposit of oxidized lipoproteins and cholesterol, unleashes an inflammatory and immune reaction, which is the first step of the atherogenic cascade. To say it in terms of the Virchovian atherogenic theory of «response to injury», the oxidized lipoproteins and the crystallized cholesterol deposited and trapped in the sub-endothelium of the arterial wall are the primeval «injury», untying an inflammatory, immune and ultimately scar repair «response». But without the deposit and entrapment of oxidized LDL-cholesterol in the vascular wall, the development of atherosclerosis is unlikely.
The Seven Countries Study (SCS), linking saturated fat to heart disease must be rejected. The Seven Countries Study (SCS),32 conducted by the also legendary US American physiologist Ancel Keys, was a seminal, observational, longitudinal, and prospect cohort study, started in 1958, which took into account diet and another lifestyle traits and their correlation with degenerative heart disease or coronary heart disease.32,33 The original chosen countries were: United States, Italy, Finland, Greece, Holland, Japan, and Yugoslavia (upon the dissolution of the latter, Croatia and Serbia composed two different nations, which explain why the study of seven countries became one of eight). But more than be a multinational study, the trial focused on selected geographic regions, comprising 16 local cohorts of men aged 40-59.34 As a whole, the study revealed a close relationship between the percentage of dietary calories derived from saturated fat and mortality from coronary heart disease.34 In the third part of this reflection, we will discuss that not all the saturated fatty acids have the same pathogenic capacity, as well as the fact that the polyunsaturated fat transformed by industrial processes in trans compounds is highly atherogenic.
Since the beginning, some critics questioned that Keys selected some nations, leaving out others, putting the US in an end and Japan in the other, constructing then an almost linear relationship between fat consumption and the rate of deaths per 100,000 inhabitants. Self-appointed prosecutors have accused Keys to cherry-picked the collected information (the selection of the best or most desirable data), and also have blamed him of fudging the numbers, or simply branded him as having committed fraud.35 The organization named True Health Initiative,34 focused in proven principles of lifestyle related to health, has written a superb White Letter that examined all the misconceptions and blatant lies of revisionists against both, the study and its main investigator, demonstrating that when the whole material is dissected, all the iconoclast’s arguments fade down. For example, the anti-Keys revisionists frequently use the graphs displayed in Figures 1 and 2 to try to show that many countries were eliminated on purpose from the SCS, in order to line up the data with the preconceived idea that fat consumption was behind the surge of atherosclerotic diseases. The first graph was exhibited in a previous Keys communication,36 using data on heart disease of the Food and Agriculture Organizations (FAO) and the World Health Organization (WHO), from six (no seven) countries.
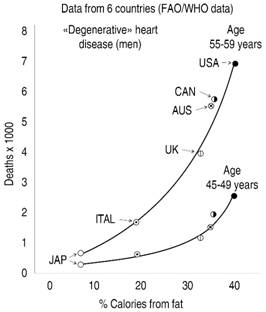
Modified from: Keys A. Atherosclerosis: a problem in newer public health. J Mt Sinai Hosp NY. 1953; 20: 118-139.
Figure 1: Relationship between percentage of calories derived from fat and mortality from «degenerative» heart disease.
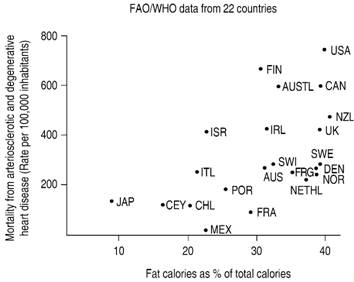
Adapted from: Yerushalmy J, Hilleboe H. Fat in the diet and mortality from heart disease: a methodologic note. N Y State Med. 1957; 57 (14): 2343-2354.
Figure 2: Mortality from degenerative and arteriosclerotic heart disease and the percentage of total calories from fats consumed by men aged 55-59 years.
Figure 2 shows the estimations of Yerushalmy & Hilleboe,37 graphing the data from 22 countries from the same source. Some revisionists conclude that correlation between fat consumption and disease is lost in this second estimation with a greater number of nations, but this is a false conclusion, as correlation remains, although not with the statistical strength of the 6 countries graph. But what is a monumental lie is to affirm, based on these two graphs, that some countries were eliminated on a tricky way, in order as not to affect the study hypothesis. To say it once, none of these data were from the SCS. While the first graph, as has been stated in previous paragraphs, pertain to a previous Keys communication, the second one was published a year before SCS started.
But indeed, the first graph, was the starting point of Keys hypothesis… «it must be concluded that dietary fat is somehow associated with cardiac disease mortality, at least in middle age».36 The participant countries were non-randomly selected, according with the White Letter´s authors,34 but instead chosen if they had reliable dietary data and vital statistics. In 1952, for example, Mexico lacked this pertinent information, or the available one was not well validated. In other instances, some nations were discarded because they still suffered the stragglers of World War II and the food shortages of the immediate postwar period. In addition, some countries declined to participate, because they lack financial or technical resources, or because the contacted researchers were not interested in the study. This latter was the explanation of the exclusion of France, and not a purportedly decision to eliminate it, because the «French Paradox»38 phenomenon (the contradictory observation that France has a low incidence of coronary heart disease, despite the consumption of a great quantity of saturated fat), would hurt the trial hypothesis. Moreover, the concept of that paradox was enunciated several years after the starting of the trial, so it was impossible that could influence the selection or not of France.38 This false argument (the tricky exclusion of France) is employed very frequently by the revisionists.
Figure 3 shows the relation between the percentage of calories derived from saturated fat and mortality rates from coronary heart disease, in 16 «ecological» regions comprised in the study.34 The study showed that the consumption of saturated fat was the main determinant of serum cholesterol concentration and not, the dietary cholesterol itself. So, the distortion of dietary counselling, which then took place, limiting drastically, and somehow irrationally, the cholesterol consumption, was not derived from the results of the SCS nor from any specific recommendation of the same Keys.

Figure 3: Mortality from coronary heart disease (CHD). Seven Countries Study. U.S. raildoad (A); Belgrade, Servia (B); Crevalcore, Italy (C); Dalmatia, Croatia (D); East Finland, Finland (E); Corfu, Greece (G); Crete, Greece (K); Montegiorgio, Italy (M); Zutphen, Holland (N); Slavonia, Croatia (S); Rome, Italy (R); Tanushimaru, Japan (T); Ushibuka, Japan (U); West Finland, Finland (W); Velika, Krsna, Serbia (V); Zrenjanin, Serbia (Z).
Finally, another accusation is in regard to the correlation between sugars and heart disease. As the mentioned White Paper commissioned by The True Health Initiative34 states, there is a popular sense claiming «that prevailing nutrition science is incorrect». A not insignificant number of serious authors and vulgar bloggers think that it is the excess of carbohydrates in the habitual diet of the contemporary human being, more than the saturated fat, the responsible fact of the surge of heart disease. The SCS researchers indeed were interested in stablish the link between sugar intake and heart disease, and in fact, they proved a meaningful statistical association, which disappear when fat income was introduced in the analysis.34,39-41 Anyhow, it is not possible to stop considering the contribution of the high ingestion of carbohydrates, and its consequence, prolonged hyperglycemia as a very important coadjutant factor in the genesis of atherosclerosis, mainly in diabetic patients. Several mechanisms of vascular damage caused by hyperglycemia had been established.42 Probably, the most important pathogenic mechanism is related to the overproduction of AGEs, (advanced glycated end-products), formed by non-enzymatic oxidation of glucose and proteins. These products, interacting with a selective AGE transmembrane receptor of the immunoglobulin superfamily (RAGE),43 cause an activation of nitroxidative stress cascade, an inflammatory reaction and an overexpression of several growth factors activators, as protein kinase C (PKC). This and other mechanisms of hyperglycemia explain the extended and long-lasting vascular and organic damage in the chronic elevation of blood glucose. The relation between high ingestion of carbohydrates and atherosclerosis in general population, is less striking than in diabetics, and merits a broader discussion. In the third section of this text we will deepen even more in this subject.
In the long time that has passed since the results of the SCS were published to date, a voluminous set of data provided by several evidence sources have reinforced the general conclusion of SCS. In the section devoted to the «diet iconoclasts» we will address again with greater depth this dietary issue.
Statins have not eradicated coronary heart disease. Many critics of the cholesterol hypothesis cited the overoptimistic and little cautious statement of Nobel Prize laureates (for the discovery of the gene which codifies the LDL receptor) Goldstein and Brown:44…« exploitation of recent breakthroughs-proof of the cholesterol hypothesis, discovery of effective drugs, and better definition of genetic susceptibility factors-may well end coronary disease as a major public health problem early in the next century». as an example that the hypothesis is a blatant failure, because that wishful thinking simply did not come true. Of course, atherosclerosis is a multifaceted lesion caused by a constellation of rather complex biological precursors and environmental determinants. And although blown-up LDL hypercholesterolemia is the main cause of atherosclerosis, it is hardly the only one. To say it once, if LDL-C does not reach an anti-natural, non-physiologic, concentration, atherosclerosis does not develop clinically in humans, or experimentally in animals, as it was established in multiple experimental, anthropological, epidemiological, and interventional observations, already commented.
There are a handful of reasons explaining why hypolipidemic treatments, while they have improved the prognosis of many patients, especially those at high risk, evidently have not attained the unlikely goal of making atherosclerosis disappear from human pathology. Among the reasons that prevent the «eradication» of atherosclerosis by hypolipidemic interventions, to begin with, is the fact that the accepted goals of lowering C-LDL are insufficiently strict. It is often forgotten or left aside the datum that the physiologic LDL cholesterol serum concentration of human being is below 70 mg/dL, in the vicinity of the Figure of 50 mg/dL, according with our ape forage-eater lineage. So, even a «low» level of LDL of 100 mg is about 40-50% higher than the normal physiological concentration. The «normal» values of total (< 200 mg/dL) or LDL cholesterol (70-100 mg/dL) represents truly a somehow moderate dyslipidemic level. In this context it is important to remember the index called cumulative cholesterol exposure45 (grams of cholesterol per year) over a lifetime, which explains that the development of atherosclerosis is the result of both, the magnitude of hypercholesterolemia plus the duration of the exposure time. A non-dimensional value of this index around 6 is a threshold for the occurrence of atherosclerotic events. So, a patient with familial homozygous hypercholesterolemia having 800 mg/dL of serum total cholesterol, at the end of the tenth year of age will have a cumulative cholesterol exposure of 8, while a person with a «normal» total cholesterolemia of 180 mg/dL, would need around 45 years of exposure to attain the same threshold. Associated conditions like other cardiovascular risk factors can lessen the threshold, while the absence of them or the female gender puts it higher. If a person has true physiologic total cholesterol concentration of 100-120 mg/dL, as it happened in many Paleolithic men and is observed in both, wild apes and contemporary hunter-gathering tribesmen,46 it would be necessary 70-80 years to reach the atherogenic threshold.
Another probable cause against the disappearance of atherosclerosis by statin influence is the condition colloquially called «closing the barn door after the horse is out», English expression that means that taking precautions after the event has already happened, has no value at all. A similar phrase in Spanish, more dramatic but with the same admonitory meaning is: «cover the well after the child is drowned». In cardiovascular prevention this concept means that interventional reduction of LDL-cholesterol comes too late, in primary prevention, because treatment is in general initiated in adulthood (sometimes in advanced ages) and not during youth, and in secondary prevention, obviously, because the reduction treatments are started just after the clinical explosion of a vascular disease, which before the acute event, was already present, but in a long asymptomatic stage. As it is known, the process of developing atherosclerotic lesions, since early fat streaks (atherosis) to complicated, vulnerable or calcified plaques, can take a very long time. Many of the immature fatty lesions can regress, but the more advanced atheromatous fibro-lipid lesions, with fibrotic, necrobiotic, or calcification components are unlike to disappear, no matter how intensive the lipid-lowering regime could be.
Another question that interferes with the usefulness of cholesterol reduction in the abolishment of atherosclerosis is the residual risk, as it is named the threat that remains after a low concentration of LDL-C is attained with proper treatment. Among the various mechanisms related to this phenomenon is the atherogenic effect of other lipoproteins containing apo B100 apolipoprotein, like VLDL and IDL or the decrease of the protective HDL-cholesterol. Although the role of triglycerides in atherogenesis has been questioned,47 there is considerable evidence that various types of hypertriglyceridemia (those associated to insulin resistance and/or with apolipoproteins containing apo B100 or apo E) are related to vascular complications.48 Although an excess of chylomicrons does not cause atherosclerosis, when the concentration of VLDL and IDL particles rises, there is an increase in small and dense LDL (strong predictor of coronary atherosclerotic disease), more attracted to the sub-endothelial proteoglycans and sulfated glycosaminoglycans, and easier to be oxidized. Although therapies directed to increase HDL values have failed to demonstrate an important decrease of vascular risk, it is undeniable the role that hypoalphalipoproteinemia plays in this context.49 Then, the single correction of LDL cholesterol is not enough to attenuate the risk impact of the entire dyslipidemic spectrum.
In clinical grounds, just to mention an example, the effect of severe cholesterol disturbance, as happens in patients with homozygous familial hypercholesterolemia, who experience myocardial infarctions and coronary deaths at early age, without any other vascular risk factor, is a powerful argument favoring the cholesterol hypothesis.50 In the heterozygous variety of familial hypercholesterolemia, as well as in the more numerous cases of high-risk patients with true intolerance to statins or veritable proof that standard treatment is unable to reach therapeutic goals, the relatively new PCSK9 monoclonal antibodies inhibitors (evolocumab and alirocumab), not only represent a novel strategy, but also confirm the cholesterol hypothesis.51 In the very high-risk individuals, PCKS9 inhibitors have not reduced importantly all-cause mortality on top of the statin effect, but have induced some kind of coronary lesion regression. For example, in the Glagov study,52 the addition of a PCSK9 inhibitor in patients treated with statins decreased the atheromatous volume 5.8 mm3 with evolocumab while this reduction was only of 0.9 mm3 with placebo. PCSK9 inhibitors in comparison with the combination of statins and ezetimibe, decreased LDL-C by 39.20%, accompanying this decline with a concomitant reduction of 1.06% of cardiovascular risk, according with a recent Cochrane review.53 Could revisionists exhibit robust data of this extension and quality contradicting the cholesterol hypothesis?
Lowering cholesterol levels does not improve mortality rates. Several recent meta-analyses have shown the reduction of both, all-cause and cardiovascular mortality, as well as the lower incidence of cardiovascular events, in all kind of patients, including those without previous vascular complications and in absence of severe dyslipidemia. As anticipated, the greatest benefits occurred in the highest risk patients.54 In the study of the Cholesterol Treatment Trialists,55 which included 174,000 patients from 27 randomized trials, for every downsizing of LDL cholesterol of 1 mol (38.6 mg/dL) the risk of major vascular events was reduced 21%. In subjects with low risk (less than 10%), each 1 mmol/L reduction in LDL cholesterol was associated with a reduction of about 11 per 1,000 of major vascular events in a lapse of 5 years. All-cause mortality was reduced 10%, and the relative risk of cardiovascular events, about 20-25%. These Figures were similar across the entire patient’s spectrum: on primary or secondary prevention, at all risks, and in both genders. In addition, a recent comprehensive statement from the European Atherosclerosis Society Consensus Panel,56 show hardly disputable facts about the certainty of the cholesterol hypothesis, as a number of genetic studies and therapeutic randomized intervention trials have demonstrated that lowering of LDL-C causes a proportional reduction of the rate of atherosclerotic events. The pooled data from numerous essays (Mendelian randomization studies, n = 194,427 subjects) and interventional studies (randomized controlled trails and prospective cohort studies, with the combined N of more than 600,000 individuals) show a log-linear correlation between LDL serum concentration and the risk of cardiovascular disease. Several prestigious trails, whose data had been used in this super-analysis, were TNT, PROSPER, CARE, JUPITER, ALLHAT-LLD, WOSCOPS, ASCOT LLA, PROVE-IT and IMPROVE-IT, among others, representing the talent and efforts of many world-class researchers. In a recent meta-analysis57 comprising 312,175 patients from 49 trials, using several therapies based on up-regulation of hepatic LDL receptor expression (statins, bile acid sequesters, and intestinal by-passes), a significant reduction of major coronary events was demonstrated with the proportional decrease of LDL cholesterol levels. In other recent meta-analysis,58 in the setting of secondary prevention, again it was observed a proportional reduction in the occurrence of major vascular events with the decrease of LDL-C, not only with statins, but also with the addition of ezetimibe, or PCSK9 inhibitors. In primary prevention, several meta-analyses have shown also a decrease of mortality with statin therapy in patients with defined cardiovascular risk factors.59 Furthermore, in a recent Cochrane meta-analysis considering eighteen randomized control trials in primary prevention, enclosing 56,934 participants,60 it was estimated a reduction of 14% in all-cause mortality achieved by statin treatment. A statistically significance reduction of fatal and no fatal cardiovascular disease, ischemic heart disease, stroke and revascularization procedures was also shown, without serious undesirable effects.
If the criteria of Bradford Hill are applied to judge the causal relationship between hypercholesterolemia and risk56,61 it is clear that all causality criteria are fully met: plausibility, strength, biological gradient and temporal sequence, specificity, consistence, coherence and reduction of risk with therapeutic interventions. As the European Consensus Statement declares, all the available evidence indicates «a remarkably consistent dose-dependent log-linear association between the absolute magnitude of exposure to LDL-C and the risk of ASCVD, and together demonstrate that the effect of LDL-C on the risk of ASCVD increases with increasing duration of exposure».56
The conclusive lesson of all this is that we are using wrongful cut-points and diagnosing hypercholesterolemia too late, that is, we are closing the well curb extemporarily, when the child is already drowned or at least, agonizingly kicking in the water. A collage made mixing the closing remarks of the European document with some samples of clinical wisdom gives us invaluable arguments to oppose the iconoclastic clash: 1) the cumulative arterial burden (magnitude of the disorder plus time) foretell the initiation and progression of atherosclerosis. 2) the preventive motto: the more the better, signals the importance of the maximal possible reduction of LDL-C concentration to obtain the maximal clinical and preventive results, leading to the second and complementary saying: cholesterol concentration, the lower the better, completely true, because for each 1% percent of reduction of the lipoprotein there is a 1% diminishing of atherosclerotic risk. In other words, the international overwhelming experience (seminal controlled studies and gigantic meta-analyzes) establishes undoubtedly that the cholesterol hypothesis is currently an accurate scientific statement. 3) In that context, the descent of both, relative and absolute risk, are proportional to the magnitude of the reduction of LDL-cholesterol level.
Lessening cholesterol concentrations in the elderly is not useful. High cholesterol serum concentrations help survival. It is known that although cardiovascular disease is a major cause of mortality and morbidity in the elder, physically active aged people has, in general, lower rates of cardiovascular diseases,62 but at the same time, the majority of aged people, mainly those affected by frailty, face a high probability of suffer cardiovascular events.63
Because there are a handful of confounders derived from the heterogeneity of elderly populations, and the effect in morbidity and mortality of other diseases and conditions, often seen in advanced ages, the smallest and weakest body of evidence about the cholesterol hypothesis comes from older age groups, who, in general, are not well represented in many lipid-lowering trials. Due also to the natural shorter life expectancy in final years, there is a lack of certainty regarding the usefulness of statin treatment to improve life expectancy in this age group.64 Although there are few studies including only the elderly population, several trials that encompass older groups, along with other of younger ages, show in general a benefit in the reduction of the risk of cardiovascular events in those aged people subjected to a LDL reduction intervention.65 Although several studies have shown, in general, a similar benefit in every age stratum,66,67 in the Heart Protection Study (HPS),68 using simvastatin, a medium-strength statin, the risk of major cardiovascular outcomes decreased more in patients aged 65 years than in younger people. Likewise, a post-hoc analysis of the Greek study GREACE,65 made evident that statin treatment reduced both relative and absolute cardiovascular risk to a greater degree in aged people than in younger subjects.
Aside from these facts, the outcome of three main trials (PROSPER,69 JUPITER70 and HOPE-371), threw different and somehow contradictory results. In PROSPER study (using pravastatin, a weak statin), a composite final point (composed by coronary death, nonfatal myocardial infarction, and fatal or nonfatal stroke) was not reduced by statin therapy, although mortality from coronary disease went down 24% in the pravastatin treated group. Instead, in JUPITER trial, the composite end point (formed by myocardial infarction, unstable angina, stroke, arterial revascularization or cardiovascular death) was dropped by rosuvastatin (a powerful statin), although all-cause mortality did not decreased. Finally, in the HOPE-3 trial, using also rosuvastatin, in the group of patients aged 65 year or more, the composite end point (cardiovascular death and nonfatal myocardial infarction or stroke) was descended 25% with statin treatment. In old persons aged 80 years or more, there are also conflicting conclusions.72 While a group of studies did not demonstrate any benefit in these oldest subjects, or even a higher mortality with the lowest concentrations of total cholesterol, a meta-analysis showed an evident diminishing of mortality (mainly in men), proportional to the decreased concentration of total cholesterol.73
A recent meta-analysis, limited to studies written in English language and informed only in PUBMed, led by Ravnskov,74 found that elevated concentrations of LDL-C were inversely related to mortality in people over 60 years of age. In this work, many studies were rejected for a number of reasons, including those that seemed «irrelevant» (sic) to the authors, judging the relevance only with the reading the abstracts, and leaving only 19 studies, enclosing 30 cohorts, and 68,094 subjects. Apart of the fact that this author has been one of the main obsessive flag bearers of the anti-cholesterol crusade, to the point of having sent more than 50 rebuttal letters or commentaries against the cholesterol hypothesis to many journals, his analysis had been found faulty and misleading. Some of the methodological flaws were «a lack of understanding of major national health guidelines, of selection and survivor bias, adjustment inclusions and exclusions, and of consideration of statin use».75 At any rate, we believe that meta-analyses, including those that are well planned and executed, do not provide better scientific information than large randomized, controlled trials.
Besides, to begin with, no every person is affected equally by the same risk factor. For example, although epidemiologically smoking is related to lung cancer, this outcome affect only about 20% of men and 25% of women per 100,000 person-years.76 In such a way, tobacco is doubtlessly carcinogenic, but its effects are not the same in all people. It may be that these smoker persons, who reach an old age and never had or will have lung cancer, belong to that class of individuals, that for some genetic reason are resistant to the carcinogenic effects of tobacco. In this sense, there must be a certain segment of the population that for some reason is resilient to the atherogenic effect of mild hypercholesterolemia, probably via an immunomodulatory action on oxidized LDL. In younger populations, these «cholesterol-immune» persons are not many numerous, and in consequence do not affect the general cholesterol level association with atherosclerotic vascular events. But the amount of these cholesterol resistant survivors increases proportionally to the number of the already deceased persons affected by atherosclerotic diseases, who were, as the majority of humans, prone to develop atherosclerosis related to mild or severe LDL-hypercholesterolemia. It is also true that cholesterol concentrations raise gradually from adolescence until the mature age,77 after which it is observed a descent in both genders, probably due to a reduced hepatic biosynthesis of cholesterol caused in turn by the decline of organic functionality of old age.78 But also, this population with low cholesterol concentrations may be the result of a «survival bias»,78 in which persons with naturally lower cholesterol levels live longer than hypercholesterolemic individuals, incrementing in such way the proportion of lower population concentration of the lipid.
Furthermore, we have some insight about the genetic resistance to cholesterol, at least in animal models. Certain laboratory rat strains (Sprague-Dawley, Wistar-Furth, Spontaneously Hypertensive, Fischer 344)79 show a very little or any increase in cholesterol levels when nourished with high cholesterol content chow. There are experimental evidence that the excess in dietary cholesterol is compensated by a marked reduction of de novo cholesterol hepatic synthesis and an increment of free cholesterol bile elimination. But in this regard, human beings function in the same way as these rats, being intestinal absorption of dietary cholesterol a less important determinant of cholesterolemia.
In conclusion, the greater strength of the available evidence points to the fact that the preventive effect of LDL reduction is similar or higher in advanced ages. This hardy disputable fact cannot be denied by the results of a single meta-analysis plagued of a number of methodological flaws.

