










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de cardiología
versión impresa ISSN 0188-2198
Rev. Mex. Cardiol vol.26 no.3 México jul./sep. 2015
Review
Pheochromocytomas: diagnosis and treatment
Feocromocitomas: diagnóstico y tratamiento
Reinaldo Alberto Sánchez-Turcios*
* Endocrinologist and MSc Pharmacology. Hospital General Milpa Alta, México, D.F.
Correspondence to:
Reinaldo Alberto Sánchez-Turcios
Tepic Núm. 113-610,
Col. Roma Sur,
Del. Cuauhtémoc, 06760, México, Distrito Federal, México.
Tel. 015552648061
Cel. 0445543508824
E-mail: rturcios@live.com.mx
Recibido: 27/07/2015
Aceptado: 25/08/2015
ABSTRACT
Pheochromocytomas are neoplasms that have their origin in chromaffin cells of the adrenal medulla. 80 to 90% of these are located in one of the adrenal glandules. This pathology is characterized by multiple symptoms that constitute a complex, heterogeneous clinical frame with a high rate of cardiovascular morbidity and mortality. The main secretion is catecholamine metabolites: metanephrine and normetanephrine. Diagnosis is carried out by determining free metanephrines in plasma (not conjugated) and fractioned metanephrines in 24-hour urine collection. Its location through different image procedures is fundamental. Preoperative treatment is initiated with a adrenergic antagonist and by adding, after a week, b adrenergic antagonists. Trans-operative treatment requires a multidisciplinary team of medical experts. This treatment is of vital importance and depends on the size and existence of metastasis. In some cases, adrenal retroperitoneal laparoscopy is preferred. However, an anterior approach is used when the tumor is > 6 cm, but other physicians have considered a 6 cm to 15 cm size. Transoperative follow up is a vital procedure for the patient. Paragangliomas are extra-adrenal ganglia pheochromocytomas.
Key words: Pheochromocytoma, catecholamine metabolites, surgical treatment.
RESUMEN
Los feocromocitomas son neoplasias que tienen su origen en las células cromafines de la médula adrenal; 80 a 90% están localizados en una de las glándulas adrenales. Es una patología caracterizada por múltiples signos y síntomas que constituyen un cuadro clínico heterogéneo, complejo y con alto índice de morbilidad y mortalidad cardiovascular. La principal secreción son los metabolitos de las catecolaminas: metanefrina y normetanefrina. El diagnóstico se realiza con la determinación de metanefrinas libres en plasma (no conjugadas) y metanefrinas fraccionadas en orina de 24 horas; la localización es fundamental por diferentes procedimientos de imágenes. El tratamiento preoperatorio inicialmente es con antagonistas a adrenérgicos y agregándose una semana después antagonistas b adrenérgicos. El tratamiento transoperatorio requiere de un grupo de profesionales versados en la materia. El tratamiento transoperatorio es de vital importancia. Su tratamiento actual depende del tamaño y de la existencia o no de metástasis. Se ha preferido laparoscopia adrenal vía retroperitoneal; se utiliza la vía anterior cuando el tumor es > 6 cm; otros han considerado el tamaño de 6 cm a 15 cm. Los paragangleomas son feocromocitomas de los ganglios extra-adrenales.
Palabras clave: Feocromocitoma, metabolitos de catecolaminas, tratamiento quirúrgico.
INTRODUCTION
Pheochromocytomas are neuroendocrine neoplasias originated in the chromaffin cells of the adrenal medulla. This medulla biosynthesizes and hyper-secretes catecholamine, its metabolites, and other proteins such as epinephrine, norepinephrine, metanephrine, etc. It takes its name from the brown granules (pheo) which are produced by the oxidation of the catecholamines with the chromic acid. Paraganglioma is a tumor derived by the extra-adrenal chromaffin cells of the sympathetic and paravertebral ganglia. These tumors are also found along the parasympathetic ganglia located in the glossopharyngeal and vague nerves of the neck and cranium base.1 These paragangliomas do not produce catecholamines, 80-85% of the chromaffin cell tumors are pheochromocytomas, while 15-20% of these tumors are paragangliomas (PGL).2
Catecholamines biosynthesis-metanephrines
The adrenal medulla has two cell populations that synthesize catecholamines and other proteins through intracrine, endocrine, and paracrine mechanisms. The most well-known of these is the interaction of the nicotinic receptor bonded to G proteins in the membrane. This bond develops a sequence of biochemical and physical processes to activate the enzymes: tyrosine hidroxilasa tyrosine → DOPA, decarboxilasa of the L aromatic amino acids → dopamine, β hydroxylase dopamine → norepinephrine, phenylethanolamine-N-methyltrasferase (induced by cortisol) → epinephrine, COMT (catechol-O-methyl transferasa) → normetanephrine and metanephrine mao → mopgal (3-methoxy-4-hydroxy-phenyl glycoldheyole). Pacap (pituitary adenylate cyclase-activating polypeptide) acts like a neurotransmitter that regulates the release of catecholamines and can implement trophic and apoptotic effects. These could influence the progress and differentiation of neoplasic cells. NPY (Y neuropeptide) is a peptide with a 36 amino acid chain present in the normal adrenal medulla and in the pheochromocytoma that regulates locally the secretion of catecholamines. Cortisol activates COMT enzyme, at the intra-adrenal portal between cortex and medulla, to synthetize and to secret catecholamines. The larger concentrated bio-synthetized catecholamine is epinephrine with 80% of all the catecholamines secreted by the medulla in normal conditions. Norepinephrine is the catecholamine mainly synthetized in the sympathetic ganglia.3,4
EPIDEMIOLOGY
This condition has an incidence of 2-8 cases out of 1,000,000 inhabitants a year.5 This figure has been underestimated since 50% of the pheochromocytomas were found in one series autopsies.6 The prevalence of pheochromocytomas and paragangliomas in hypertensive population varies between 0.2 to 0.6%.7-10 The condition happens at any age, but is more frequent between the 4th to 5th decades. It has the same frequency in both sexes.
PGL prevalence in children with hypertension11 is approximately 1.7%. About 5% of patients with incidentaloma have pheochromocytomas.12,13 Some PGLs are potentially malignant. Malignity is defined as the presence of metastasis of the chromaffin tissue. Their prevalence is between 10% to 17%, but it can rise to more than 40% in patients with mutations of the gen that codes the enzyme succinate dehydrogenase subunit B (SHDB).14-16
CLINICAL INDICATORS
Due to its clinical heterogeneity, this tumor has been called the "great mimic". It has the classic triad: episodic headache, generalized diaphoresis and tachycardia. About 50% of them have paroxysmal hypertension and 40% sustained systemic arterial hypertension, 5 to 15% are normotensive. Patients can present other symptoms: papilledema, dyspnea, pallor, general weakness, panic spells, orthostatic hypotension, blurred vision, papilledema, weight loss, polyuria, polydipsia, constipation, globular sedimentation speed, hyperglycemia, leukocytosis, thrombocytosis, erythrocytosis, psychiatric disorders, cardiomyopathy due to excess catecholamines, lung acute edema, arrhythmia, anesthesia induced hypertension, surgery. The 10% rule: About 10% of these tumors are malignant; 10% are present in children; 10% are bilateral; 10% are extra-adrenal; 10% are familial ones. (Hereditary pheochromocytoma).17
PHEOCHROMOCYTOMA SHOULD BE SUSPECTED IN PATIENTS WHO HAVE ONE OR MORE OF THE FOLLOWING:
• Resistant hypertension18, refractory hypertension.19• Hypotension crisis.
• Hyperadrenergic spells (episodes, palpitations, diaphoresis, headache, tremor, or pallor).
• Unfavorable cardiovascular responses to anesthesia, tricyclic antidepressants, phenothiazine, histamine etc.
• Incidentaloma adrenal.
• Genetic history of pheochromocytoma.
• Manifestation of hypertension on patients as young as 20 year old.
• Hypertension concomitant with impaired fasting glucose, impaired glucose tolerance, and diabetes mellitus.
DIAGNOSIS
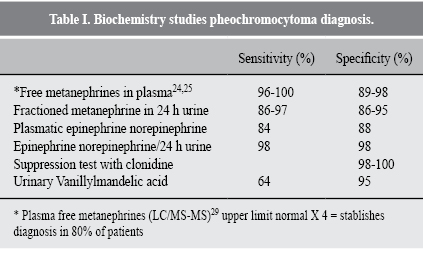
The most important biochemistry tests for diagnosing pheochromocytomas are (Table I): free plasma metanephrines determination.20,21 Free plasma normetanephrine and metanephrine, method for the measurement of plasma metanephrines using solid phase extraction-liquid chromatography- tandem mass spectrometry22 and urine of 24-hour fractionated metanephrines. Free plasma metanephrines can be used to predict the size23 and the plasmatic concentration of methoxytyramine, concomitant with normetanephrine, can determine the localization of extradrenal neoplasia.24 Free plasma metanephrines are from 12 % to 30% higher at the sitting and supine position, respectively.25
Precision diagnosis of free plasmatic metanephrines measurements, has been now confirmed by several studies.26-28
CLONIDINE SUPPRESSION TEST
Clonidine stimulates the α2 adrenergic receptors at the brain and prejunctional neuronal levels, thus if there is a decrease of elevated normetanephrine to normal concentrations after the clonidine test, indicates that the sympathetic activation is its source, and a lack of decrease in plasma free normetanephrine < 40% and the persistence of plasma free methaneprhine > 0.61 nmol/L three hours after administering clonidine indicate the presence of pheochromocytoma. This test is indicated in cases in which the elevation plasma normethanephrine is mild.
Clonidine suppression test methodology
1) It is determined by the basal concentration of free catecholamines and metanephrines in plasma.2) It is orally administered 0.3mg clonidine.
3) 3 hours later, free catecholamines and metanephrines in plasma are carried out, and it is positive if their concentrations remain high after three hours.29 This test criterion has a 90% plasmatic accuracy.
Eisenhofer et al. informed 100% of sensitivity and 96% of specificity.29
Neuropeptide Y
Plasma neuropeptide Y levels are increased 87% in patients with pheochromocytoma30 and chromogranin A in 80%.31
WHAT CAN INCREASE FREE METANEPHRINE IN PLASMA AND/OR FRACTIONED METANEPHRINE IN 24 H URINE?
Medications: Alfametildopa, tricyclic antidepressants, monoamine oxidase inhibitors, b-adrenergic receptor antagonists, acetaminophen, abrupt discontinuation of clonidine, selective β-adrenergic antagonists (doxazosin, terazosin, prazosin), calcium-channel antagonists, sympathomimetics (pseudoephedrine, amphetamines), pheonoxybenzamine, serotonin norepinephrine reuptake inhibitors, buspirone, levodopa.
Foods: coffee (caffeic acid), potatoes, fermented foods, cereals, processed meat, beans, nuts, tomatoes, fruits.
Others: Position (sitting versus supine for 20 minutes before blood draw), cigarette smoking, exercise, age > 60 years old, untreated OSA, chronic heart failure, renal dysfunction.
LOCATION TESTS (Table II)
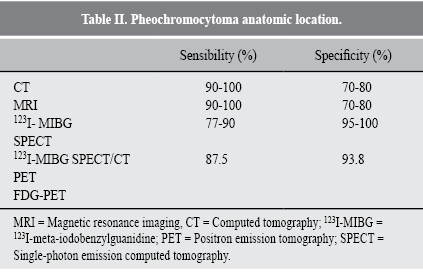
1. Computed tomography has a 10 Hounsfield attenuation units.32 Necrosis cystic growths and calcifications can be observed in tumor tissue. Tumor tissue in the range of 1.2-1.5 a media of 5.5.332. Imaging magnetic resonance (IRM) shows hyperintense images in T2. However, hemorrhages, cysts add to these tumors heterogeneity, and even 35% of these cannot exhibit a pheochromocytoma; they are frequently isointense to muscle and hypointense to liver.32
3. Use of MIBG (123I - Metaiodobenzylguanidine) which is captured by sympathomedullary system cells. The functional image can be obtained by labelling 131I and 123I.34 However, 123I is preferred due to its low radioactive dose and its short life; it does not have β emissions.34 Sensitivity is 70 to 90%, specificity 95% to 100%,32 MIGB allows total body evaluation to detect extra-adrenal neoplasias, metastasis and relapses.
4. SPECT (Single-photon emission computed tomography); It is the most adequate in transversal cuts, but it has its limits for small lesions.35,36 The 123I MIBG SPECT/CT combination has a sensitivity of 87.55% and a specificity of 93.8%.
5. PET (positron emission tomography) with FDG (fluorodeoxyglucose) has a limited sensitivity in benign tumors, but malignant tumors are easily detected.37
6. We suggest the use of positron emission tomography with 18F-fluorodeoxyglucose (18F-FDG PET/CT) in patients with metastasis disease. 18F-FDG PET/CT is the preferred image rather than gammagraphy with 123I-MIBG in patients with known metastatic pheochromocytoma and paraganglioma.
7. We suggest that patients with paragangliomas have a study for mutations with succinate dehydrogenase (SDHB), and patients with metastasis should be analyzed for mutations with SDHB.
Pre-trans- post-operatory management
Pre-operative treatment
Pre-operative treatment is the most important one. Its objective is to control arterial hypertension, tachycardia and intravascular volume contraction with a diet rich in liquids and ClNa to prevent hypotension. We use the following scheme: we initiate treatment with prazosin 1 mg every 8 hours during a week, and 2 mg every 8 hours in the second week. Immediately, we add propranolol 40 mg every 8 hours for 3 days. After this, we use 80 mg every 8 hours. With this scheme, hypertension is generally controlled. Then we infuse expansive solutions and hemotransfusions if necessary. The elective treatment is:
1. Antagonist α-adrenergic medication is the elective one. Phenoxibenzamine is a α-adrenergic antagonist non-selective, irreversible which is considered the initial medication due to its long standing action and irreversibility. It has a limited tolerance due to side effects. After administering α-adrenergic antagonist, one must add β-adrenergic antagonist for tachycardia and/or arrhythmia treatment. β1-selective antagonist are preferred; β2 non-selective antagonist could antagonize the vasodilation action of β2.2. Calcium channels antagonists can be combined with α and β1, or can be used as primary medications to control hypertension. β adrenergic stimulation in the juxtaglomerular apparatus can stimulate renin secretion that may be present in a group of patients with pheochromocytoma; then the inhibiting medication of convertasa enzyme from angiotensin I to angiotensin II, and blockers could improve arterial blood pressure control in those patients. Less toxic side effects in those patients are a useful alternative for intolerance to α-adrenergic antagonists.
3. Catecholamine synthesis inhibitors, like α-methyl-L-tyrosine inhibits tyrosine hydroxylase. Its effect is observed after three days of use. Chronic vasoconstriction produces a status of volume depression. The main objective is minimize pre-operative hypovolemia. Volume expansion must be initiated once artery hypertension, even in patients, with hemodynamic instability and/or cardiomyopathy can be considered.
Hyperglycemia is produced by an increase of glycogenesis and glycogenolysis; this is due to the stimulation of catecholamines by α and β adrenergic receptors in the hepatic tissue. The decrease of the insulin by the stimulation of the α and β-adrenergic receptors of β cell and the decrease of the glucose intake by the skeletal muscle.
Other physicians have considered the following via:
• α-adrenegic antagonists: nonselective α-antagonists: phenoxybenzamine 10-20 mg 2-3 times/day (100 mg max daily), selective α-antagonists: doxazosin 1 mg daily, terazosin 1mg 2 times/day, prazosin 1 mg 3 times/day (all 20 mg max daily).• Calcium channel antagonists: nifedipine SR 30-120 mg daily, nicardipine 30 mg 2 times/day (120 mg max daily).
• Catecholamine synthesis inhibition: metyrosine 250 mg 3-4 times/day (4 g max daily).
• Others: β-antagonists, angiotensin II converting enzyme inhibitors (ACEI), angiotensin II receptor blockers (ARB).38,39
Peri-operative management
Adrenalectomy is the management for pheochromocytomas.
Laparoscopic surgery is the standard operative treatment for pheochromocytoma excision. It can be performed via trans-abdominal (≥ 9 cm) or retroperitoneal depending on tumor size. Retroperitoneal surgery has been shown as a safer alternative to trans-abdominal laparoscopy.
Exploratory laparoscopy is indicated when malignity is suspected.
Once the patient is normotensive, hemodynamically stable, located metastasis with vascular and/or capsular invasion and the compromise to adjacent structures; surgical approach sould be analized. It is of outmost importance to have a team formed with an endocrinologist, laparoscopy surgeons, and an anesthetist. It is also necessary to keep an arterial line assessing blood pressure every minute. This is important due to the fact that trans-operative neoplasia handling produces a tumor catecholamine extrusion to the bloodstream, and a sudden rise of arterial pressure; there are even moments during intervention when a pause has to be done to control hypertension. We have used phentolamine bolus. In most of our cases (10 patients), we have use an infusion, and a close communication with intensive care unit.
We recommend minimally invasive adrenalectomy for most of adrenal pheochromocytomas. Open resection is advisable when pheochromocytomas are big (> 9 cm) to assure a complete tumor resection. We suggest partial adrenalectomy for selected patients such as hereditary pheochromocytoma, with small tumors that have already gone through a complete contralateral adrenalectomy, so that the adrenal cortex could be preserved and prevent permanent hypocortisolism.40,41
Intraoperative management
Adrenergic antagonists: phentolamine 1-5 mg IV boluses or infusion, esmolol 0.5 mg/kg over 1 minute then 0.05 mg/kg/min infusion.
Calcium channel antagonists: nicardipine 5 mg/hour infusion titratable to 15 mg/hour, clevidipine 1-2 mg/hour infusion titratable to 32 mg/hour.
Vasodilators: nitroprusside 2 μg/kg/min, not to exceed 800 μg/hour and magnesium sulfate bolus 40 mg/kg, infusion 1-2 μ/hour.42,43
Postoperative management
Adrenergic antagonists: phenoxybenzamine 10-20 mg 2-3 times/day, doxazosin 1mg daily, terazosin 1 mg 2 times/day, prazosin 1 mg 3 times/day.
Catecholamine synthesis inhibition: metyrosine 250 mg 3-4 times/day.
Postoperative follow up
Ten days after surgery, free plasma, and fractionated metanephrines in 24 hour urine must be counted. If they are normal, vigilance must be continued every six months for two years; after this, every year if normal. Patients that evolved hypertension free plasma and fractionated in 24 hour urine metanephrine count must be reassessed; a morphological search for metastasis must be performed and establish the surgical or chemotherapeutic treatment. Other patients can evolve with residual hypertension and their treatment is with minimum doses of α1, and β1 adrenergic antagonists.44,45
REFERENCIAS
1. De Lellis RA, Lloyd RV, Heitz PU, Eng C. Phatology and genetics of tumours of endocrine organs (IARC WHO classification of tumors). Third edition. Lyon, France: World Health Organization. 2004. [ Links ]
2. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005; 366: 665-675. [ Links ]
3. Schultz C, Eisenhofer G, Lehnert H. Principles of catecholamine biosynthesis, metabolism and release. Front Horm Res. 2004; 31: 1-25. [ Links ]
4. Eisenhofer G, Lenders JW, Pacek K. Biochemical diagnosis of pheochromocytoma. Front Horm Res. 2004; 31: 76-106. [ Links ]
5. Stenstrom G, Svardsudd K. Pheochromocytoma in Sweden 1958-1981. An analysis of national cancer registry data. Acta Med Scand. 1986; 220: 225-232. [ Links ]
6. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Reviw of a 50-year autopsy series. Mayo Clin Proc. 1981; 56: 354-360. [ Links ]
7. Sinclair AM, Isles CG, Brown I, Cameron H, Murray GD, Robertson JW. Secondary hypertension in a blood pressure clinic. Arch Intern Med. 1987; 147: 1289-1293. [ Links ]
8. Anderson GH Jr, Blakeman N, Streeten DH. The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens. 1994; 12: 609-615. [ Links ]
9. Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocr Pract. 2000; 6: 249-252. [ Links ]
10. Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004; 27: 193-202. [ Links ]
11. Wyszynska T, Cichocka E, Wieteska-Klimczak A, Jobs K, Januszewicz P. A single. Pediatric center experience with 1,025 children with hypertension. Acta Paediatr. 1992; 81: 244-246. [ Links ]
12. Mantero F, Terzolo M, Arnaldi G. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000; 85: 637-644. [ Links ]
13. Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev. 2004; 25: 309-340. [ Links ]
14. Plouin PF, Fitzgerald P, Rich T. Metastatic pheochromocytoma and paraganglioma: focus on therapeutics. Horm Metab Res. 2012; 44: 390-399. [ Links ]
15. Brouwers FM, Eisenhofer G, Tao JJ. High frequency of SDHB germline mutations in patients with malignant catecholamine producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006; 91: 4505-4509. [ Links ]
16. Amar L, Baudin E, Burnichon N. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007; 92: 3822-3828. [ Links ]
17. Stein PP, Black HR. A simplied diagnostic approach to pheochromocytoma. A review of the literature and report of one institution's experience. Medicine. 1991; 70: 46-66. [ Links ]
18. Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr et al. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure The JNC 7 Report. JAMA. 2003; 289 (19): 2560-2571. [ Links ]
19. Acelajado MC, Pisoni R, Dudenbostel T, Dell'Italia LJ, Cartmill F, Zhang B et al. Refractory hypertension: definition, prevalence, and patient characteristics. J Clin Hypertens (Greenwich). 2012; 14: 7-12. [ Links ]
20. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991; 40: 544-556. [ Links ]
21. Pacek K, Eisenofer G, Ahlman H. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Rev. 2007; 3: 92-102. [ Links ]
22. Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, GrebeSK, Murad MH. Pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2014; 99: 1915-1942. [ Links ]
23. Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM et al. Pheochromocy-toma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem. 2005; 51: 735-744. [ Links ]
24. Eisenhofer G, Goldstein DS, Sullivan P, Csako G, Brouwers FM, Lai EW et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab. 2005; 90: 2068-2075. [ Links ]
25. de Jong WH, Eisenhofer G, Post WJ. Dietary influences on plasma and urinary metanephrines: implications for diagnosis of catecholamine-producing tumors. J Clin Endocrinol Metab. 2009; 9: 2841-2849. [ Links ]
26. Lenders JW, Pacak K, Walther MM. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002; 287: 1427-1434. [ Links ]
27. Raber W, Raffesberg W, Bischof M. Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med. 2000; 160: 2957-2963. [ Links ]
28. Sawka AM, Jaeschke R, Singh RJ, Young WF Jr. A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab. 2003; 88: 553-558. [ Links ]
29. Eisenhofer G, Goldstein DS, Walther MM, Friberg P, Lenders JW, Keiser HR et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true-from false-positive test results. Journal of Clinical Endocrinology and Metabolism. 2003; 88: 2656-2666. [ Links ]
30. Mouri T, Sone M, Takahashi K. Neuropeptide Y as a plasma marker for phaeochromocytoma, ganglioneuroblastoma and neuroblastoma. Clin Sci. 1992; 83: 205. [ Links ]
31. Modlin IM, Gustafsson BI, Moss SF. Chromogranin A biological function and clinical utility in neuroendocrine tumor disease. Ann Surg Oncol. 2010; 17: 2427-2443. [ Links ]
32. Leung K, Stamm M, Raja A. Pheochromocytoma: The range of appearances on ultrasound, CT, MRI, and functional imaging. AJR. 2013; 200: 370-378. [ Links ]
33. Blake MA, Kaira MK, Maher MM. Pheochromocytoma: an imaging chameleon. Radiographics. 2004; 24: 87-99. [ Links ]
34. Mullins F, O'Shea P, FitzGerald R, Tormey W. Enzyme linkedimmunoassay for plasmafree metanephrines in the biochemical diagnosis of phaeochromocytoma in adults is not ideal. Clin Chem Lab Med. 2012; 50: 105-110. [ Links ]
35. Intenzo CM, Jabbour S, Lin HC. Scintigraphic imaging of body neuroendocrine tumors. Radiographics. 2007; 27: 1355-1369. [ Links ]
36. Baez JC, Jagannathan JP, Krajewski K. Pheochromocytoma and paraganglioma: imaging characteristics. Cancer Imaging. 2012; 12: 153-162. [ Links ]
37. Derlin T, Busch JD, Wisotzki. Intraindividual Comparison of I- 123 MIBG SPECT/MRI, I- mIBG SPECT/CT, and MRI for the detection of adrenal pheochromocytoma in patients with elevated urine or plasma catecholamines. Clin Nuc Med. 2013; 38: 1-6. [ Links ]
38. Bravo EL, Tagle R. Pheochromoocytoma: state-of-the-art and future prospects. Endocr Rev. 2003; 24: 539-553. [ Links ]
39. Prys-Roberts C. Phaeochromocytoma-recent progress and its management. Br J Anaesth. 2000; 85: 44-57. [ Links ]
40. Toniato A, Boschin IM, Opocher G, Guolo A, Pelizzo M, Mantero F. Is the laparoscopic adrenalectomy for pheochromocytoma the best treatment? Surgery. 2007; 141: 723-727. [ Links ]
41. Matsuda T, Murota T, Oguchi N, Kawa G, Muguruma K. Laparoscopic adrenalectomy for pheochromocytoma: a literature review. Biomed Pharmacother. 2002; 56 Suppl 1: 132s-138s. [ Links ]
42. Hariskov S, Schumann R. Intraoperative management of patients with incidental catecholamine producing tumors: a literature review and analysis. J Anaesthesiol Clin Pharmacol. 2013; 29: 41-36. [ Links ]
43. McMillian WD, Trombley BJ, Charash WE, Christian RC. Phentolamine continuous infusion in a patient with pheochromocytoma. Am J Health Syst Pharm. 2011; 68: 130-134. [ Links ]
44. Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal-time for a reappraisal? Eur J Endocrinol. 2011; 165 (3): 365-373. [ Links ]
45. Mannelli M. Management and treatment of pheochromocytomas and paragangliomas. Ann NY Acad Sci. 2006; 1073: 405-416. [ Links ]
Nota
Este artículo puede ser consultado en versión completa en: http://www.medigraphic.com/revmexcardiol

