











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de heterotaxia (SH), también denominado situs ambigous, se define como una anomalía en la disposición de los órganos respecto a sus ejes derecho-izquierdo, principalmente involucrando órganos torácicos y abdominales1. Esta entidad difiere del situs solitus y del situs inversus2. El concepto «heterotaxia» deriva del griego heteros, que significa diferente, y taxis, que significa configuración. Igualmente suele denominarse con el término de «isomerismo»3,4. Su incidencia es rara, presentándose en cada 10,000 a 20,000 recién nacidos vivos, de predominio masculino5,6.
Se ha clasificado dicho síndrome en dos tipos: isomerismo derecho (ID) o asplenia si la configuración auricular es derecha e isomerismo izquierdo (II) o poliesplenia si la configuración es izquierda. Embriológicamente, al final de la tercera semana, durante el periodo de diferenciación de las células cardiacas y a partir del nódulo de Hensen, se lleva a cabo la lateralidad de los órganos. En pacientes con SH se ha visto la expresión génica de ciertos genes como: Nodal, Pitx2, NKX2-5, CRELD1, LEFTY2, ZIC3 y CRIPTC. Se asocian a la codificación de la vía del factor de crecimiento transformante beta. Al verse alterada dicha vía, se determina una de las dos entidades: dextromorfismo o levomorfismo7,8.
En el ID, las estructuras bilaterales presentarán características morfológicas derechas, como, por ejemplo, aurícula derecha bilateral. Es posible la presencia de ciertos defectos cardiacos tales como la trasposición de grandes arterias, válvula auriculoventricular común, hipoplasia ventricular, atresia pulmonar y obstrucción de venas pulmonares. En el isomerismo auricular derecho se carecerá de aurícula izquierda, lo que impedirá recibir el drenaje venoso pulmonar. Generalmente se asocian con cardiopatías cianóticas graves en la infancia, incluso representa una causa de mortalidad durante el primer año de vida, en aquellos casos donde hay atresia pulmonar y anomalías de retorno venoso pulmonar6,9,10.
Por su parte, en el II existe un mal desarrollo de las estructuras del lado derecho, en donde presentarán características morfológicas izquierdas como bronquios izquierdos bilaterales, pulmones bilobulados, ausencia de nódulo sinoauricular. Además, suele acompañarse a menudo con poliesplenia. Las anomalías cardiovasculares más frecuentes son las no cianóticas como defectos del cojín endocárdico, tabique auricular y en el 50% se pueden a asociar con dextrocardia6,9,11.
Se debe enfatizar un diagnóstico prenatal temprano de la entidad, ya que suele estar asociada a anomalías cardiacas complejas hasta en un 4%. De la misma forma, se deben identificar las anomalías extracardiacas, para abordarlas en la etapa posnatal (Tabla 1). Dada la complejidad del SH, su diagnóstico representa un gran reto para el ginecoobstetra y el médico materno fetal, pero una vez identificado, se debe derivar oportunamente al servicio de cardiología pediátrica12. Las patologías cardiacas requieren un manejo posnatal especializado, en su gran mayoría quirúrgico, lo cual tiene un impacto en el pronóstico a largo plazo de esta entidad7. Aun aquellos defectos aislados se deben protocolizar, debido a que pueden verse implicadas algunas morbilidades como malrotación intestinal, atresia biliar, desórdenes respiratorios e inmunitarios2.
Tabla 1 Anomalías cardiacas y otras malformaciones asociadas al isomerismo derecho e izquierdo
| Características | Isomerismo derecho | Isomerismo izquierdo |
|---|---|---|
| Malformaciones cardiovasculares | Aurícula única con apéndices auriculares derechos bilaterales | Apéndices auriculares izquierdos bilaterales |
| Mesocardia/dextrocardia | Ventrículos desequilibrados | |
| Ventrículo derecho único | Drenaje venoso pulmonar anómalo | |
| Ventrículo derecho de doble salida | Vena cava inferior izquierda persistente que drena hacia la aurícula izquierda | |
| Discordancia auriculoventricular | ||
| Arco aórtico del lado derecho | ||
| Mala posición de las grades arterias | ||
| Estenosis/atresia pulmonar | ||
| Venas cavas superiores bilaterales | ||
| Otras malformaciones/disfunciones | Pulmones y bronquios bilaterales del lado derecho | Pulmones y bronquios bilaterales del lado izquierdo |
| Bronquio corto | Bronquio largo | |
| Pulmones trilobulados | Pulmones bilobulados | |
| Asplenia | Poliesplenia | |
| Hígado simétrico | Hígado en línea media | |
| Estómago del lado derecho | Atresia/hipoplasia biliar extrahepática | |
| Malrotación intestinal | Atresia de la vena porta extrahepática | |
| Malrotación intestinal |
Se ha reportado que el diagnóstico prenatal oportuno tiene un impacto positivo en los resultados quirúrgicos y menor mortalidad perinatal. Sin embargo, al tratarse de una entidad de difícil diagnóstico, esta representa un reto en la identificación de defectos anatómicos y fisiológicos, que ayuden a planear un manejo quirúrgico preciso12,13.
En el presente artículo se expone un caso diagnosticado de forma temprana durante el ultrasonido del primer trimestre, el cual se asoció a una malformación cardiaca.
Presentación del caso clínico
Paciente de sexo femenino de 42 años, secretaria, originaria y residente de Huejotzingo, Puebla. Antecedentes familiares de diabetes mellitus tipo 2 por padre y madre. Pareja de 45 años, de oficio carpintero, sin antecedentes familiares ni personales de relevancia.
Cursa su tercera gestación. En la primera gestación se obtuvo un recién nacido masculino vivo, vía abdominal a la semana 34 por restricción del crecimiento fetal. En su segunda gestación presentó aborto a la semana 12, al estudio genético se diagnosticó feto con trisomía 18 (síndrome de Edwards). Durante la presente gestación, permaneció hospitalizada por amenaza de aborto en la séptima semana de gestación, egresando sin complicaciones.
Acudió a control prenatal para ecografía de tamizaje del primer trimestre (11-13.6 semanas) durante la semana 12 de gestación. Se realizó una ecografía en donde se corroboró vitalidad fetal y se objetivó una biometría fetal acorde a la edad gestacional. Al estudio anatómico se obtuvo una medición de la translucencia nucal aumentada de 3.7 mm (Fig. 1), se apreció la burbuja gástrica situada en el lado derecho y los demás órganos dispuestos en situs contrario, observándose una imagen en espejo (Figs. 2 y 3).
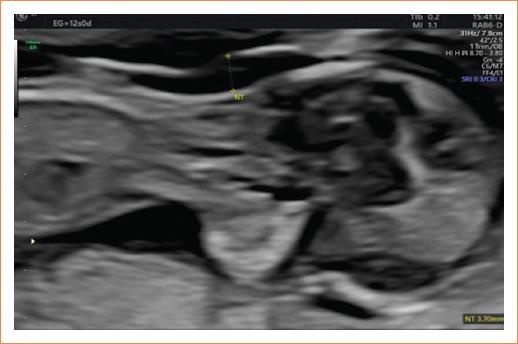
Figura 1 Feto con translucencia nucal aumentada de 3.7 mm a las 12 semanas de gestación, el cual es un marcador mayor en el primer trimestre. Se deben descartar cromosomopatías y cardiopatías asociadas.
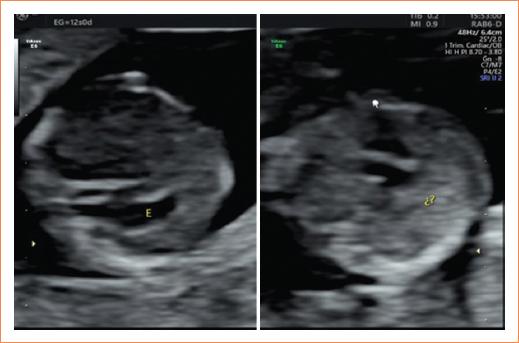
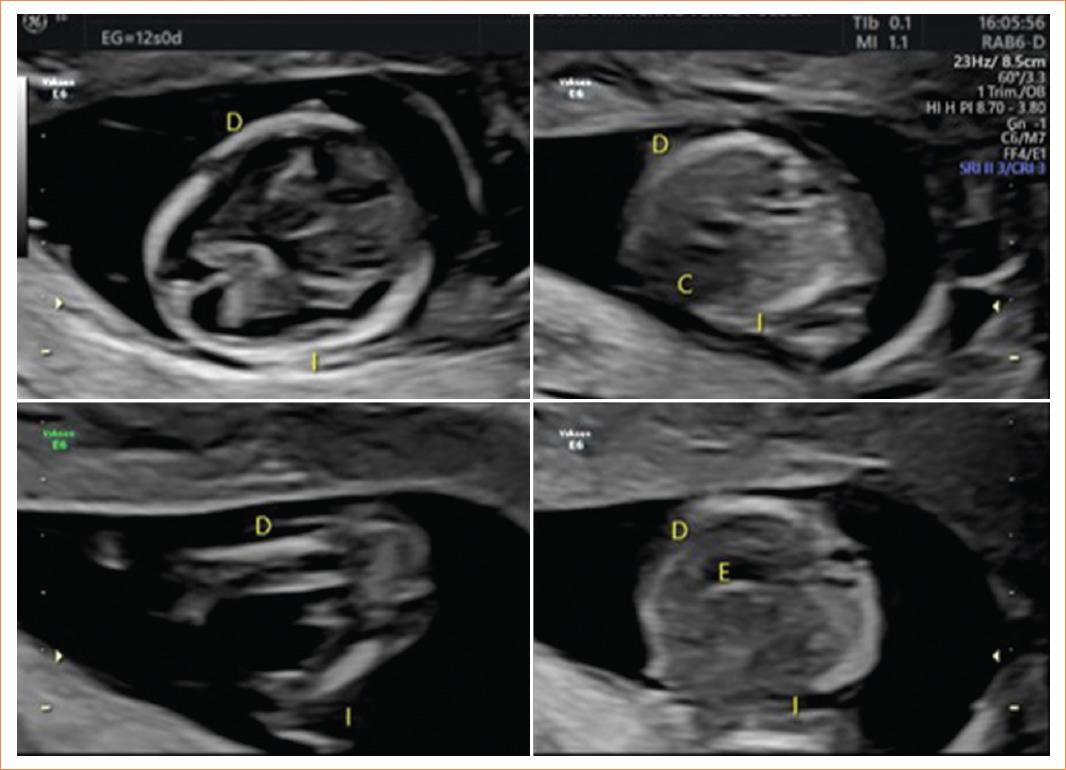
Figura 3 Situs ambiguous, se pueden apreciar la imagen «en espejo» y la disposición de los órganos en el situs contrario.
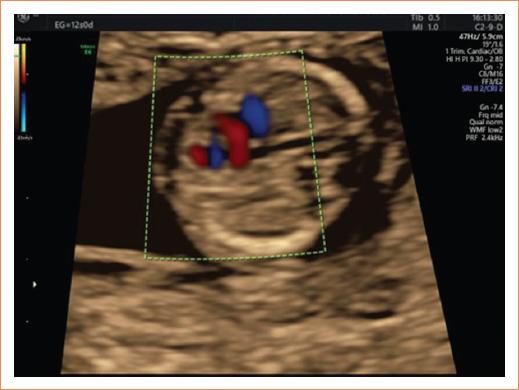
En la evaluación cardiaca, se presentó tendencia a la taquicardia fetal y a la aplicación del Doppler color se visualizó una cardiopatía asociada, tratándose de una comunicación interventricular (Fig. 4). Se optó por amniocentesis a la semana 16 para realizar examen genético, descartándose cromosomopatías. Se decidió vigilancia ecográfica estrecha en tercer nivel y se propuso intervención quirúrgica posterior al nacimiento, para corrección quirúrgica de cardiopatía congénita.
Discusión
Históricamente, en 1955 se describió por primera vez el SH por Ivemark, quien lo detalló como un «síndrome teratológico de simetría visceral» asociado a defectos cardiacos conotruncales14. Posteriormente, en 1962, van Mierop y Wiglesworth constataron que se trataba de una entidad más compleja, que no únicamente se asociaba a defectos cardiacos, sino que además se asociaba a anomalías esplénicas, pulmonares y hepáticas9,15.
Durante el desarrollo embrionario, entre la quinta y sexta semana de gestación, se produce una asimetría derecha-izquierda fisiológica en los órganos toracoabdominales, lo cual hace que tengan una correcta disposición. Existen cuatro etapas embriológicas importantes, donde se determina el situs correcto de los órganos: 1) ruptura de la simetría izquierda-derecha en la embriogénesis temprana; 2) transmisión de señales del nodo al mesodermo de la placa lateral; 3) expresión genética asimétrica de la placa lateral izquierda del mesodermo, y 4) morfogénesis asimétrica izquierda-derecha de los órganos6.
La nomenclatura fue establecida con referencia al ápex cardiaco. Existen dos tipos de isomerismo, el izquierdo que se asocia a poliesplenia y el derecho con asplenia. La poliesplenia tiene una incidencia de uno de cada 10,000 nacidos vivos, mientras que la asplenia tiene una frecuencia de uno en cada 20,000 nacidos vivos13.
La ecocardiografía fetal ha permitido realizar diagnósticos tempranos para así poder derivar a centros cardiológicos pediátricos y planear una intervención multidisciplinaria. El equipo debe estar integrado por médicos materno-fetales, cardiólogos pediatras y cirujanos pediatras, para así poder optar por un manejo en la vida posnatal temprana con el fin de mejorar el pronóstico y supervivencia de esta patología13,16. En el presente caso, se realizó un diagnóstico temprano, incluso antes que la edad gestacional promedio reportada en la literatura médica, que es alrededor de la semana 24 de gestación.
A la ecografía, existen características particulares que pueden determinar si se trata de un ID o II, aunque no en todos los casos se sigue un patrón clásico, lo que dificulta su diagnóstico prenatal6.
La característica diagnóstica más útil para determinar un II es la interrupción de la vena cava inferior en su segmento hepático, así como la continuación que tiene con la vena ácigos en el segmento distal. De la misma forma, puede estar presente un bloqueo cardiaco aurículo-ventricular, en donde clínicamente estos fetos pueden cursar con bradicardia17. Con la modalidad Doppler color, en una vista longitudinal, la vena ácigos y la aorta pueden verse de lado a lado. En caso de que exista un bloqueo cardiaco completo, este puede ocasionar insuficiencia cardiaca, hidrops fetal y muerte intrauterina9.
El diagnóstico ecográfico del ID es más complejo. El dato ecocardiográfico que puede apoyar su diagnóstico es la ausencia del seno coronario y vena cava superior, aunque no es una característica patognomónica del ID. Otra anomalía frecuente es el drenaje anómalo de las venas pulmonares9.
Además, es imprescindible realizar una evaluación radiológica minuciosa de cada órgano y sistema, para identificar anormalidades anatómicas que puedan comprometer la función y a su vez ofrecer un manejo óptimo18. En el caso que se presenta, se logró realizar el diagnóstico de una comunicación interventricular con apoyo de la modalidad del Doppler color. Su diagnóstico temprano permite ofrecer un manejo certero en la vida posnatal.
Teele et al. han reportado que existen factores cardiacos y no cardiacos que repercuten en la supervivencia. En cuanto a los factores cardiacos, los que han demostrado mayor impacto en el pronóstico son el isomerismo atrial izquierdo vs. isomerismo atrial derecho. Aquellos casos de II que no son tratados han demostrado tener una mayor supervivencia, ya que el 20% de los pacientes sobrevivirá a la infancia, ya que tienen menores efectos hemodinámicos. La mayoría de los pacientes con ID, aproximadamente el 50-100%, suelen presentar ventrículos únicos, lo cual disminuye la supervivencia10.
Otro factor de riesgo importante es la anatomía de las venas pulmonares. Alrededor del 75% de los fetos con SH tendrán conexiones pulmonares anormales, especialmente aquellos con un ventrículo único10.
Por otra parte, existen factores de riesgo extracardiacos, como los pulmonares, gastrointestinales e infecciosos. En cuanto a los defectos pulmonares, se debe corroborar la discinesia ciliar a nivel pulmonar, ya que algunos infantes pueden requerir de traqueostomías tras las intervenciones quirúrgicas. A nivel gastrointestinal, cerca del 40-90% presentará malrotación intestinal y deben intervenirse quirúrgicamente. En algunos casos puede presentarse la malformación de Abernethy, comprendida por hipertensión pulmonar, enfermedad hepática, malformación pulmonar arteriovenosa, representando un pobre pronóstico. Igualmente, el SH puede cursar con asplenia, lo que incrementa el riesgo de infecciones. Se documenta que del 21 al 30% de los pacientes con SH puede presentar sepsis10.
En cuanto al abordaje de la resolución de la gestación, Lee et al. hacen hincapié en que esta debe ser interrumpida posterior a la semana 39, ya que se ha documentado que aquellas gestaciones que son interrumpidas entre la semana 37 y 38 se asocian a un mayor riesgo de muerte perinatal posterior a las intervenciones quirúrgicas en la vida posnatal13.
Actualmente, el pronóstico ha mejorado gracias a las intervenciones oportunas, siendo la supervivencia a un año del 79-88% y a cinco años del 73-81%. El isomerismo izquierdo suele presentar un mejor pronóstico posnatal que el isomerismo derecho, dado que este último se asocia con malformaciones cardiacas graves13.
Conclusiones
El diagnóstico del SH resulta complejo, dadas las diferentes variedades de presentación que tiene. En algunas ocasiones se diagnostica de manera causal, debido a la gran asociación que tiene con las malformaciones cardiacas y abdominales.
Es necesario tener una vasta compresión de la embriología para realizar una correcta interpretación y descripción de la morfología en los patrones ecográficos. Se puede tomar gran ventaja de la ecografía para realizar un diagnóstico oportuno de las malformaciones tanto cardiacas como extracardiacas.
El diagnóstico durante la gestación permite ofrecer un manejo óptimo, principalmente en cuanto a los defectos cardiacos, posterior al nacimiento. El abordaje debe involucrar a un equipo multidisciplinario, que realice una correcta protocolización, la cual impactará positivamente en el pronóstico si se realiza de manera temprana.

