










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkPerinatología y reproducción humana
versión On-line ISSN 2524-1710versión impresa ISSN 0187-5337
Perinatol. Reprod. Hum. vol.28 no.4 Ciudad de México oct./dic. 2014
Caso clínico
Tumores de ovario de la granulosa. Informe de casos y revisión de la literatura
Granulose ovarian tumors. Case report and literature review
José Martín Palacios-Acosta,* Angélica León-Hernández,‡ Amador Ramírez-Reséndiz,§ Vanessa Carrasquel-Valecillos,II Jaén Echávez-del Riego,II Daniel Hernández-Arrazola¶
* Jefe de Servicio de Cirugía Oncológica. Instituto Nacional de Pediatría.
‡ Médico General, UNAM-FES Iztacala.
§ Médico adscrito de Cirugía Oncológica. Hospital de la Niñez Oaxaqueña.
II Médico Residente de Cirugía Oncológica. Instituto Nacional de Pediatría.
¶ Médico adscrito de Cirugía Oncológica. Instituto Nacional de Pediatría.
Correspondencia:
Dr. José Martín Palacios Acosta
Servicio de Cirugía Oncológica,
Instituto Nacional de Pediatría.
Av. Insurgentes Sur Núm. 3700-C,
Col. Insurgentes Cuicuilco,
04530, México, D.F.
Tel.: 1084-0900 ext. 1322
E-mail: drjpalacios@hotmail.com
Recibido: 08 de abril de 2014
Aceptado: 29 de junio de 2014
RESUMEN
Los tumores malignos de ovario son raros en la edad pediátrica y pueden originarse de los componentes del tejido ovárico; el tumor más frecuente es el de células germinales. La variedad de tumores del estroma incluye los tumores de células de la granulosa (teca y los tumores de células de Sertoli-Leydig); este grupo de tumores son raros; sin embargo, son propensos a presentarse con pubertad precoz. Se presentan tres casos clínicos de pacientes que cursaron con manifestaciones clínicas de pubertad precoz y tumoración abdominal. Se estableció un protocolo de estudio para su abordaje y, finalmente, se realizó resección quirúrgica, con evolución satisfactoria.
Palabras clave: Tumor de ovario, pubertad precoz, tumores de la granulosa, resección quirúrgica.
ABSTRACT
Malignant ovarian tumors are rare in children. They may originate from components of the ovarian tissue; the germ cell tumor is the most common. The variety of stromal tumors include granulosa cell tumors (theca and Sertoli-Leydig cell tumors); this group of tumors is rare but they are likely to present with precocious puberty. We report three cases of patients with clinical manifestations of an abdominal tumor in association with precocious puberty; we also describe the way they were approached, until final surgical resection. We had excellent results.
Key words: Ovarian tumor, precocious puberty, granulosa, tumors, surgical resection.
INTRODUCCIÓN
Los tumores malignos de ovario son raros en la edad pediátrica y pueden originarse de los componentes del tejido ovárico. Los más frecuentes son los de células germinales.1 Los tumores del estroma incluyen a los tumores de las células de la granulosa (teca y los tumores de las células de Sertoli-Leydig); este grupo de tumores son raros, pero suelen presentarse con pubertad precoz.2,3 Los tumores del estroma representan del 2 al 5% de todas las lesiones malignas del ovario.4
Los tumores de la célula de la granulosa (TCG) se dividen en dos subgrupos basados en la presentación clínica y características histológicas: TCG juvenil y TCG adulta. La mayoría de las pacientes son adultas, pero 5% son prepuberales. Las formas juveniles se diagnostican en mujeres menores de 30 años. La edad del diagnóstico de la variedad juvenil tiene dos picos de incidencia: entre los 8 y 9 años de vida, y entre los 13 y 17 años.5,6 Del 70 al 90% de las enfermas menores de ocho años de edad con este tipo de tumor, tienen signos de pubertad precoz isosexual. En el examen físico, pueden tener desarrollo de glándulas mamarias, sangrado vaginal, incremento del vello púbico e incremento en la edad ósea. En pacientes mayores, pueden presentar actividad hormonal con irregularidades menstruales y, raramente, signos de virilización.
En todas las enfermas, se encuentra masa palpable, dolor y distensión abdominal. En el 10% de los casos puede existir ruptura, y el primer síntoma es dolor abdominal como manifestación de un cuadro de abdomen agudo. En el 10% de los individuos hay ascitis.7,8
Los TCG son bilaterales en el 3% de los sujetos; en la mayoría de las pacientes, se limitan al ovario al momento del diagnóstico. Característicamente, estos tumores son de grandes dimensiones, en promedio, de 12 cm de diámetro; pueden tener componente sólido o ser completamente quísticos. De acuerdo con su historia natural, estas neoplasias tienen un crecimiento lento y recurrencias tardías.9,10
El diagnóstico es clínico y se sospecha en mujeres con datos de pubertad precoz. El diagnóstico se corrobora con estudios de gabinete, como ultrasonido (USG) y tomografía axial computarizada (TAC). Los estudios de laboratorio incluyen perfil hormonal, inhibina, alfa fetoproteína y gonadotrofina β coriónica. El tratamiento es la resección quirúrgica del tumor; sin embargo, en estadios avanzados, se puede requerir del uso de quimioterapia.11,12
CASOS CLÍNICOS
Caso 1
Femenina de seis años de edad que acudió al Servicio de Urgencias por sangrado transvaginal, acompañada de crecimiento de glándulas mamarias y la presencia de vello púbico. A la exploración física, tuvo un peso de 20 kg y talla de 119 cm, con la presencia de crecimiento bilateral de glándulas mamarias Tanner III (Figura 1), sin adenomegalias. El abdomen se encontró blando depresible y se detectó una masa abdominal palpable móvil, de consistencia renitente, de aproximadamente 7 x 8 cm, no adherida a planos profundos (Figura 2). El resto de la exploración, normal.


Se realizó USG, encontrándose un tumor con ecogenicidad mixta (Figura 3). La TAC mostró un tumor dependiente de ovario derecho con dimensiones de 6 x 9 x 9 cm de diámetro, con patrón mixto (Figura 4). Se realizó la determinación de alfa fetoproteína, con resultado de 2 μg/L; la fracción β de la gonadotrofina coriónica fue de 1 mUI/mL; la hormona luteinizante (LH), de 6 mUI/mL; la hormona folículo estimulante (FSH) fue de 7.2 mUI/mL; prolactina, 15.9 ng/mL; estradiol, 2.6 pg/mL, y la 17-OH PROG (17-hidroxiprogesterona), de 0.5 ng/dL.


Se decidió realizar resección quirúrgica, encontrando tumor dependiente de ovario derecho multiquístico (Figura 5). Se efectuó protocolo para tumores de ovario, con toma de muestras de líquido peritoneal, peritoneo anterior, epiplón y peritoneo adosado al diafragma. No se obtuvo muestra de ovario contralateral por no apreciar lesión macroscópica; no se efectuó biopsia de ganglios por no existir evidencia de lesión. Se realizó salpingooforectomía derecha.

El reporte de patología fue de tumor de la teca de ovario derecho sin lesión residual, biopsias negativas. La evolución fue satisfactoria, sin complicaciones postquirúrgicas. En el seguimiento de la paciente, se encontró involución de los caracteres sexuales secundarios en un lapso de tres meses, con un perfil hormonal dentro de parámetros normales.
Caso 2
Femenina de seis años de edad; inició su padecimiento al presentar crecimiento de glándulas mamarias, presencia de vello púbico y sangrado transvaginal. Acudió con facultativo, quien efectuó el diagnóstico de pubertad precoz e inició tratamiento hormonal, sin mejoría. Posteriormente, se realizó USG abdominal, el cual reportó lesión quística en hemiabdomen inferior, motivo por el cual se derivó al Instituto Nacional de Pediatría (INP).
A la exploración física de ingreso, se encontró peso de 22 kg, talla de 121 cm, crecimiento de ambas glándulas mamarias Tanner II (Figura 6), galactorrea, abdomen blando depresible con tumor abdominal móvil de consistencia renitente, no adherido a planos profundos (Figura 7). El resto de la exploración normal.


Se inició protocolo de estudio. El USG mostró tumor con ecogenicidad mixta (Figura 8); la TAC abdominopélvica evidenció una masa dependiente de ovario con características mixtas, quísticas y sólidas de 15 x 20 cm de diámetro (Figura 9). Los estudios de laboratorio indicaron alfa fetoproteína de 1.6 μg/L, fracción β de la gonadotrofina coriónica de 1.4 mUI/mL; LH, 6.2 mUI/mL; FSH, 7.4 mUI/mL; prolactina, 16.3 ng/mL; estradiol, 2.1 pg/mL, y 17-OH PROG, 0.6 ng/dL.


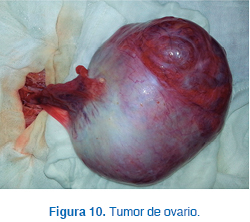
Se indicó la realización de laparotomía exploradora, en la que se encontró un tumor de ovario izquierdo de 15 x 12 cm (Figura 10). Se estableció protocolo para tumores de ovario. Posteriormente, se llevó a cabo ooforectomía sin salpingectomía más omentectomía, no habiendo complicaciones.
El reporte de patología informó el hallazgo de un tumor de células de la granulosa de ovario izquierdo, sin lesión residual y biopsias negativas. La paciente evolucionó satisfactoriamente, con involución de los caracteres sexuales secundarios en un lapso de dos meses. El perfil hormonal final se encontró dentro de parámetros normales.
Caso 3
Femenina de 12 años de edad; inició su padecimiento seis meses atrás con distensión y tumor abdominal, pérdida de peso de 10 kg, ataque al estado general, fiebre y galactorrea de dos meses de evolución.
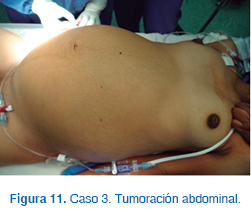
A la exploración física, tenía un peso de 26 kg, talla de 138 cm, crecimiento de ambas glándulas mamarias Tanner II, galactorrea, abdomen blando depresible con tumor abdominal móvil (Figura 11), vello púbico y resto de la exploración normal.
La TAC abdominopélvica evidenció un tumor intraabdominal heterogéneo (sólido y quístico) con densidades de 12 y 54 UH, sin calcificaciones, de 29 x 24 x 12 cm y peso de 4.61 kg (Figura 12). Los estudios de laboratorio mostraron alfa fetoproteína de 1.8 μg/L, fracción β de la gonadotrofina coriónica de 1.2 mUI/mL; LH, 6 mUI/mL; FSH, 7 mUI/mL; prolactina, 1,230 ng/mL (valor normal de referencia 0-20 ng/mL); estradiol, 2.1 pg/mL, y 17-OH PROG, 0.5 ng/dL.

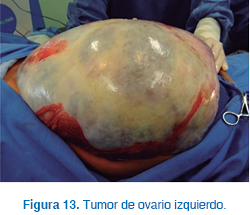
Se decidió efectuar laparotomía exploradora, en la cual se encontró un tumor de ovario izquierdo (Figura 13). Se llevó a cabo ooforectomía izquierda y protocolo para tumores de ovario, con toma de muestra de líquido peritoneal, peritoneo anterior, epiplón, peritoneo adosado al diafragma, así como toma de muestra del ovario contralateral y apendicectomía profiláctica, sin complicaciones.
El reporte de patología señaló el hallazgo de tumor de ovario de células de la granulosa de 29 cm sin infiltración en la cápsula, epiplón con inflamación crónica granulomatosa con células gigantes multinucleadas de tipo cuerpo extraño y congestión capilar, apéndice cecal con hiperplasia linfoide folicular, biopsia de peritoneo con inflamación. La paciente evolucionó satisfactoriamente, apreciándose involución de caracteres sexuales secundarios en un lapso de tres meses y prolactina de 18 ng/mL, con perfil hormonal normal.
DISCUSIÓN
La pubertad pseudoprecoz se relaciona con tumores de ovario; de ellos, hasta 60% de los casos corresponden a tumores de la granulosa; el resto consisten en cistoadenomas, gonadoblastomas, carcinomas, arrenoblastomas, tumores de las células lipoides, tecomas y quistes de ovario.13 Este grupo de tumores pueden producir crecimiento mamario y de vello púbico. La presentación clínica en la variante juvenil de los tumores de la granulosa se ha asociado con síndrome de Potter, anormalidades congénitas, enfermedad de Ollier (encondromatosis múltiple), síndrome de Maffucci (enfermedad de Ollier y hemangiomas).14 Aproximadamente 90% de los TCG se encuentran en pacientes prepuberales, aunque se han reportado casos raros de secreción de andrógenos, causando el desarrollo de virilización o hirsutismo. La persona puede percibir dolor abdominal localizado o dolor pélvico.14,15 La tríada clásica es tumor anexial palpable, niveles séricos de estradiol elevados y ausencia o disminución de gonadotrofinas.16
Este grupo de tumores se puede manifestar como abdomen agudo hasta en el 10% de los casos, de manera secundaria a ruptura o torsión del tumor. Pueden dar metástasis por vía hematógena a pulmón, hígado y cerebro. Se ha encontrado que cerca del 8% de las enfermas desarrolla metástasis por vía linfática. Los hallazgos radiológicos en la edad pediátrica son lesiones quísticas multiloculares o unilocular. El efecto estrogénico puede manifestarse con un útero alargado y engrosamiento de la mucosa endometrial e, incluso, hemorragia.17,18 Los TCG secretan estradiol, lo cual, en presencia de las células de la teca, tiene la propiedad de secretar androstenediona, con la subsecuente conversión a estradiol por las células de la granulosa, por lo que los niveles séricos de estradiol están elevados al momento del diagnóstico.19 Algunos autores han propuesto el uso de los niveles de estradiol para el seguimiento de las pacientes, y su uso para el monitoreo clínico de la enfermedad.20,21
La inhibina es un marcador que se sintetiza por las células de la granulosa y puede ser relacionada con la carga tumoral y la respuesta a la quimioterapia. Los niveles séricos de FSH y LH suelen ser bajos, y en aquellos casos de pseudopubertad temprana, se ha encontrado que los niveles de alfa fetoproteína se encuentran elevados. El antígeno carcinoembrionario C125 es un marcador sensible para los tumores epiteliales de ovario; sin embargo, no es específico, ya que se puede elevar en otros procesos intraperitoneales; no obstante, en los casos de TCG suele encontrarse elevado.22,23
El tratamiento quirúrgico tradicional se inicia con una incisión media o Pfannsteell; se debe tomar líquido peritoneal para enviar a citología. En caso de no existir líquido, se instilan 5 mL de solución salina hacia la pelvis y en los espacios paracólicos y se extrae la muestra para el análisis. En caso de que la persona presente enfermedad bilateral (2 a 8% de los casos), se debe tratar de realizar salvamento de tejido ovárico; sin embargo, en caso de enfermedad unilateral, debe llevarse a cabo la salpingooforectomía para disminuir el riesgo de recidiva. Debido a la baja incidencia de enfermedad bilateral, la biopsia del ovario contralateral es un tema controvertido y, en general, se prefiere no realizarla para preservación de la fertilidad de la paciente. El resto de la rutina de ovario se realiza de forma habitual.24
En caso de mujeres con tumores de grandes dimensiones, pueden realizarse resecciones parciales en combinación con quimioterapia. La recurrencia puede presentarse en cavidad peritoneal, hígado, pulmón y hueso. Los factores pronósticos incluyen el estadio del tumor, el tamaño y los grados de atipia nuclear que presenta, así como la actividad mitótica. La ruptura del tumor no se considera factor de mal pronóstico. En pacientes con estadio IA, no se requiere quimioterapia, incluso en aquellos casos que se han complicado con ruptura transoperatoria.25
La supervivencia para las enfermas en estadio I va desde 78 hasta 92%, y para estadio IA es del 83 al 98%. Kalfa y colaboradores26 revisaron 40 casos pre- y postpuberales, de los cuales 17 fueron diagnosticados con base en precocidad periférica. Todas las niñas que fueron diagnosticadas con TGC con base en pubertad precoz tuvieron una clasificación FIGO IA, sin presentar evidencia de diseminación peritoneal.26,27
Algunos autores han informado que cuando se retrasa el diagnóstico de tumores de la TCG en presencia de signos endocrinos, puede encontrarse diseminación peritoneal del tumor y hay un índice alto de recurrencia. Sin embargo, el pronóstico suele ser bueno, con curación cercana al 80% de los casos y con supervivencia a 4.5 años hasta en el 97% de las pacientes.
Muchos de estos tumores presentan recurrencia durante el primer año después del diagnóstico inicial, pero puede haber recurrencias tardías que se presentan hasta después de cuatro años.9,28 La recurrencia que se presenta de forma temprana puede llevar a la persona a la muerte en un lapso de 13 a 16 meses.29
Aunque no existe un consenso para las enfermas postoperadas, nosotros hacemos el seguimiento, solicitando laboratorios, estradiol, carcinoma 125 y estudios de gabinete a nivel pélvico y abdominal. A nuestras pacientes se les da un seguimiento con citas de valoración cada tres meses por un periodo de dos años, y posteriormente cada seis meses hasta completar un lapso de 10 años.
CONCLUSIÓN
Los tumores de TCG del ovario son una de las causas poco frecuentes de pubertad precoz de tipo periférico. La piedra angular en el tratamiento es la resección quirúrgica, y es necesaria para el diagnóstico histopatológico, la estadificación y, en ocasiones, para disminuir la carga tumoral. En aquellas pacientes en que se considera que el tumor es irresecable, se les puede ofrecer la oportunidad de quimioterapia. Cuando la enfermedad se detecta en estadios tempranos, se considera que tiene un pronóstico bueno. El uso de la quimioterapia se reserva para estadios avanzados y enfermedad metastásica.
En nuestra opinión, este tipo de tumores de la teca-granulosa debe ser tratado con resección quirúrgica completa, que debe incluir a la salpinge, y realizar, en forma previa, la toma de marcadores tumorales y en el tiempo quirúrgico, la llamada rutina de ovario.
REFERENCIAS
1. Adachi T, Sato H, Nakayama S, Shimizu S, Matsui H, Nakabayashi M. Primary amenorrhea due to juvenile granulosa-cell tumor of the ovary: a case report. J Obstet Gynaecol Res. 2012; 38: 597-600. [ Links ]
2. Von Allmen D. Malignant lesions of the ovary in childhood. Semin Pediatr Surg. 2005; 14: 100-5. [ Links ]
3. Fleming N, Nanassy J, Lawrence S, Black A. Juvenile granulosa and theca cell tumor of the ovary as rare cause of precocious puberty: case report and review of literature. J Pediatr Adolesc Gynecol. 2010; 23: e127-e31. [ Links ]
4. Sekkate S, Kairouani M, Serji B, Tazi A, Mrabti H, Boutayeb S et al. Ovarian granulosa cell tumors: a retrospective study of 27 cases and a review of the literature. World J Surg Onc. 2013; 11: 1-6. [ Links ]
5. Young R, Dickersin R, Scully R. Juvenile granulosa cell tumor of the ovary. A clinicopathological analysis of 125 cases. Am J Surg Pathol. 1984; 8: 575-9. [ Links ]
6. Merras L, Vettenranta K, Möttönen M, Heikinheimo M. Ovarian granulosa cell tumors in childhood. Pediatr Hematol Oncol. 2002; 19: 145-56. [ Links ]
7. Vassal G, Flamant F, Caillaud JM, Demeocq F, Nihoul C, Lemerle J. Juvenile granulosa cell tumor of the ovary in children: a clinical study of 15 cases. J Clin Oncol. 1988; 6: 990-5. [ Links ]
8. Nigil N, Agarwal G, Pandey R, Dabadghao P. Juvenile granulosa cell tumor presenting as isosexual precocious puberty: a case report and review of literature. Indian J Endocrinol Metab. 2013; 17: 157-8. [ Links ]
9. Powell J, Otis C. Management of advanced juvenile granulosa cell tumor of the ovary. Gynecol Oncol. 1997; 64: 282-4. [ Links ]
10. Outwater E, Wagner B, Mannion C, McLarney J, Kim B. Sex cord-stromal and steroid cell tumors of the ovary. Radiographics. 1998; 18: 1523-46. [ Links ]
11. González E, Fernández C, Salas J, González C. Tumor de células de la granulosa tipo adulto de ovario. Prog Obstet Ginecol. 2008; 51: 593-604. [ Links ]
12. Gershenson D. Management of ovarian germ cell tumors. J Clin Oncol. 2007; 25: 2938-43. [ Links ]
13. Schulin C, Yamamoto M, Pires Y, Mayerson D, Cattani A. Pubertad precoz periférica por tumor ovárico de las células de la granulosa juvenil, con manifestaciones iso- y heterosexuales en una niña de 6 años. Rev Med Chile. 2003; 131: 71-6. [ Links ]
14. Metwalley K, Elsers D, Farghaly H, Abdel-Lateif H, Abdel-Kader M. Precocious puberty secondary to a mixed germ cell-sex cord-stromal tumor associated with an ovarian yolk sac tumor: a case report. J Med Case Reports. 2012; 6: 162-5. [ Links ]
15. Sehouli J, Drescher F, Mustea A, Elling D, Friedmann W, Kühn W et al. Granulosa cell tumor of the ovary: 10-years follow-up data of 65 patients. Anticancer Res. 2004; 24: 1223-30. [ Links ]
16. Lack E, Perez-Atayde A, Murthy A, Goldstein M, Crigler J, Vawter G. Granulosa theca cell tumors in premenarchal girls: a clinical and pathologic study of ten cases. Cancer. 1981; 48: 1846-54. [ Links ]
17. Ko S, Wan Y, Ng S, Lee T, Lin J, Chen W et al. Adult ovarian granulosa cell tumors: spectrum of sonographic and CT findings with pathologioc correlation. Ajr Am J Roentgenol. 1999; 72: 1227-33. [ Links ]
18. Kim SH, Kim SH. Granulosa cell tumor of the ovary: common findings and unusual appearances on CT and MR. J Comput Assist Tomogr. 2002; 26: 756-61. [ Links ]
19. Lappohn R, Burger H, Bouma J, Bangah M, Krans M, De Brujin W. Inhibin as a marker for granulosa-cell tumors. N Engl J Med. 1989; 321: 790-3. [ Links ]
20. Rey R, Lhomme C, Marcillac I, Lahlou N, Duvillard P, Josso N et al. Antimullerian hormone as a serum marker of granulosa cell tumors of the ovary: comparative study with serum alpha-inhibin and estradiol. Am J Obstet Gynecol. 1996; 174: 958-65. [ Links ]
21. Cronje H, Niemand I, Bam R, Woodruff J. Granulosa and theca cell tumors in children: a report of 17 cases and literature review. Obstet Gynecol Surgery. 1998; 53: 240-7. [ Links ]
22. Kdous M, Hachicha R, Gamoudi A. Early isosexual precocious pseudopuberty revealing a juvenile granulosa cell tumor in a six-year-old girl. Gynecol Obstet Fertil. 2004; 32: 311-4. [ Links ]
23. Boggess J, Soules M, Goff B, Greer B, Cain J, Tamimi H. Serum inhibin and disease status in women with ovarian granulosa cell tumors. Gynecol Oncol. 1997; 64: 64-9. [ Links ]
24. Plantaz D, Flamant F, Vassal G, Chappuis J, Baranzelli M, Bouffet E et al. Granulosa cell tumors of the ovary in children and adolescents. Multicenter retrospective study in 40 patients aged 7 months to 22 years. Arch Fr Pediatr. 1992; 49: 793-8. [ Links ]
25. Pectasides D, Pectasides E, Psyrri A. Granulosa cell tumor of the ovary. Cancer Treatment Rev. 2008; 34: 1-12. [ Links ]
26. Kalfa N, Ecochard A, Patte C, Audran F, Pienkowki C, Thibaud E at al. A nationwide study of granulosa cell tumors in pre- and postpubertal girls: missed diagnosis of endocrine manifestations worsens prognosis. J Pediatr Endocrinol Metab. 2005; 18: 25-31. [ Links ]
27. Frausto S, Geisler J, Fletcher M, Sood A. Late recurrence of juvenile granulosa cell tumor of the ovary. Am J Obstet Gynecol. 2004; 191: 366-7. [ Links ]
28. Malmstrom H, Hogberg T, Risberg B, Simonsen E. Granulosa cell tumors of the ovary: prognostic factors and outcome. Gynecol Oncol. 1994; 52: 50-5. [ Links ]
29. Abell M, Holtz F. Ovarian neoplasms in childhood and adolescence. Tumors of non-germ cell origin. Am J Obstet Gynecol. 1965; 93: 850-66. [ Links ]
Nota
Este artículo puede ser consultado en versión completa en: http://www.medigraphic.com/inper

