










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkPerinatología y reproducción humana
versión On-line ISSN 2524-1710versión impresa ISSN 0187-5337
Perinatol. Reprod. Hum. vol.19 no.2 Ciudad de México jun. 2005
Artículos originales
Análisis y resultados clinicocitogenéticos de fetos y recién nacidos con alteraciones cromosómicas durante un año en el Instituto Nacional de Perinatología
Analysis of clinical and cytogenetic studies fetus and new born with chromosomal anomalies during one year at the Instituto Nacional de Perinatología
Monica Aguinaga,a Isabel Llano,a Rocío Báez ,a Clara Hernández,a Javier Castro,a Guadalupe Razo,a Ana Aparicio,a Conrado Uría,a José Luis Saucedo,a Juan Carlos Ibáñez,a Ricardo Meléndez,a Ma. Jesús Zavaleta,a Dora Gilda Mayén–Molina,a Ricardo García–Cavazosb
a Departamento de Genética Subdirección de Investigación Biomédica. Instituto Nacional de Perinatología.
b Encargado de la Dirección de Enseñanza del INPer.
Correspondencia:
Dra.Mónica Aguinaga Ríos.
Segundo piso.Torre de lnvestigación. Departamento de Genética. Instituto Nacional de Perinatología.
Montes Urales 800, Col. Lomas de Virreyes
C.P. 11000, México, D.F.
Tel: 5520–9900Ext. 316.
Correo electrónico: monicaguinaga@aol.com
Recibido: 22 de febrero de 2005
Aceptado: 6 de septiembre de 2005
RESUMEN
Introducción: Las anormalidades cromosómicas son una causa frecuente de morbilidad y mortalidad en la población humana.
Objetivo: Describir el número y tipo de las alteraciones cromosómicas numéricas y estructurales detectadas en estudios citogenéticos realizados prenatalmente y en recién nacidos en el Instituto Nacional de Perinatología (INPer) durante el periodo comprendido de enero a diciembre del 2003.
Metodología: Realizar un estudio descriptivo de tipo retrolectivo de los pacientes revisados por el Departamento de Genética con defectos congénitos que presentaron anormalidades cromosómicas.
Resultados: Durante el año 2003, 3.26% (189/5795) de los pacientes nacidos en el INPer presentaron defectos al nacimiento, de los cuales veintisiete pacientes mostraron un cariotipo anormal de los cuales 21 (77.7%) presentaron alteraciones cromosómicas numéricas; además, en seis (22.2%) se encontró una alteración cromosómica estructural, lo que representa 0.46% de los pacientes nacidos en el INPer. En seis casos el diagnóstico se realizó en etapa prenatal y se corroboró al nacimiento.
Conclusiones: La mayoría de alteraciones cromosómicas se presentan con múltiples defectos al nacimiento y con alteración en el crecimiento y desarrollo mental. Es importante que ante la presencia de pacientes con múltiples defectos mayores estructurales se sospechen este tipo de alteraciones y se realicen los estudios necesarios a la familia para poder brindar un adecuado asesoramiento genético.
Palabras guía: Cromosoma, cariotipo, trísomía, monosomía.
ABSTRACT
Introduction: Chromosomal anomalies are a frequent cause of human disease.
Objective: Describe the numerical and structural chromosomal anomalies detected by cytogenetic studies done prenatally and in newborns found in the Instituto Nacional de Perinatología during the period between January and December 2003.
Methods: Descriptive study of the patients with congenital defects seen by the Genetics Department who presented chromosomal anomalies.
Results: During the year 2003, the 3.46% (189/5795) of the babies born at the INPer had structural anomalies. Twenty patients had a chromosomal anomaly of which 21 (77.7%) had a chromosomal numeric alteration and six (22.2%) a chromosomal structural anomaly which represented 0.46% of the newborns. In six cases the diagnosis was done prenatally and confirmed at birth.
Conclusions: Most of the chromosomal anomalies present themselves with multiple congenital anomalies and retarded growth and development. It is very important to implement this type of studies in patients with congenital anomalies, complete the familiar study and provide an accurate genetic counseling to the parents.
Key words: Karyotype, chromosome, monosomy, trisomy.
INTRODUCCIÓN
Aproximadamente 2 a 3% de los recién nacidos vivos y 6 a 9% de los óbitos presentan algún tipo de defecto congénito.1 Estos defectos son causa de morbilidad y mortalidad en el periodo perinatal y en la edad pediátrica y contribuyen a 30–50% de las admisiones en hospitales pediátricos.2 Las causas de los defectos al nacimiento son variables, una de las principales son las alteraciones cromosómicas numéricas y estructurales.
Los cromosomas fueron vistos y nombrados por primera vez en el siglo XIX. En 1959, Lejeune fue el primero en descubrir un cromosoma extra en los pacientes con síndrome de Down.3 En ese mismo año, Ford, et al.* informaron acerca de la existencia de pacientes con un desarrollo sexual anormal y talla baja con cariotipo 45,X. La citogenética ha evolucionado y actualmente es una disciplina bien desarrollada y establecida.
Las anormalidades cromosómicas contribuyen en forma importante a la morbilidad y mortalidad en el periodo perinatal. Muchas de ellas, si bien permiten la sobrevida del producto, causan un retraso en el desarrollo psicomotor y retraso mental del mismo. El desbalance cromosómico se detecta en aproximadamente 0.3% de todos los recién nacidos, en 4% de los óbitos y en 50 a 60% de los abortos del primer trimestre.5,6 Las anomalías más frecuentes son los rearreglos estructurales balanceados (4.3%), en donde no existe pérdida o ganancia del material genético, los que a su vez se clasifican en translocaciones, inversiones e inserciones. En las alteraciones desbalanceadas, sí existe una ganancia o pérdida del material genético, ya sea de un cromosoma completo (aneuploidia) o de parte de éste. Las más frecuentes son las alteraciones de los cromosomas sexuales (3.9%), las trisomías autosómicas de los pares 13,18 y 21 (1.4%), y los rearreglos estructurales (0.7%). Con excepción de ciertas aneuploidias sexuales, la mayoría de estas alteraciones producen retraso mental y defectos congénitos mayores y menores específicos para cada anomalía. Más aún, las trisomías 13 y 18 son consideradas letales al nacimiento. En productos provenientes de abortos, son frecuentes otro tipo de aneuploidias, como la trisomía 16, monosomía del X y triploidias (presencia de tres complementos haploides), entre otras.7
El estudio citogenético prenatal se ofrece a las pacientes con alguno de los siguientes antecedentes: edad materna avanzada, hijo previo con cromosomopatía, antecedentes de pérdida gestacional recurrente, parejas con rearreglo cromosómico estructural, tener un resultado anormal de triple marcador sérico y la presencia de defectos estructurales por ultrasonido.8
Los cromosomas se distribuyen a cada célula hija durante la división celular en un proceso preciso, aunque pueden ocurrir errores en la segregación de los cromosomas, ocasionalmente existe tendencia a distribuciones erróneas. La mayoría de las alteraciones cromosómicas ocurren por un proceso de no–disyunción durante la meiosis I o II, lo que ocasiona un gameto cromosomicamente anormal. Aproximadamente una de cada 625 personas es portadora de una translocación recíproca,9 el rearreglo puede ocurrir como un evento de novo o puede ser transmitido por alguno de los padres.
Durante la meiosis I, el apareamiento de los homólogos se encuentra alterado cuando se forma el cuadrivalente, durante paquiteno cada segmento se aparea con su homólogo. La distribución de los homólogos hacia los extremos se conoce como segregación, existen cuatro patrones básicos de segregación (Figura 1). En la figura no se muestra la segregación 4:0, ya que no se ha observado en recién nacidos vivos.
Los defectos estructurales causados por aneuploidias cromosómicas son secundarios a un desbalance en la dosis génica, la cual se encuentra disminuida en los casos de monosomía o deleción o incrementada en trisomía o duplicación. Dos hipótesis principales han sido consideradas como posible causa de los defectos estructurales congénitos y del retraso mental observado en pacientes con alteraciones cromosómicas. La primera denominada "reduccionista" o "aditiva", se refiere a la acción directa de los genes en el segmento o cromosoma involucrado.10 La segunda, llamada "inestabilidad en el desarrollo", se basa en que el fenotipo entre las diferentes aberraciones cromosómicas es similar y que el imbalance génico amplifica la inestabilidad inherente en varias características epigenéticas involucradas en el desarrollo.11
En ocasiones, el estudio citogenético convencional no es suficiente para reconocer el segmento del cromosoma involucrado debido al tamaño del segmento, por lo que se deben realizar técnicas de citogenética molecular como la hibridación in situ con fluorescencia (FISH, por sus siglas en inglés), en la cual, por medio de sondas marcadas con colorantes fluorescentes hibridan un segmento específico o todo el cromosoma, pudiéndose determinar el origen del rearreglo.12
La detección de una anormalidad cromosómica es mayor en el diagnóstico prenatal, debido a la diferente sobrevida durante el embarazo; ya que un embarazo anormal tiene mayor grado de pérdida espontánea, que uno normal.13
El objetivo de este estudio es dar a conocer el abordaje de los fetos y recién nacidos con defectos congénitos cuya causa es secundaria a una alteración cromosómica numérica o estructural, en el Instituto Nacional de Perinatología, durante el periodo comprendido de enero a diciembre del 2003. El conocimiento de la causa de este tipo de defectos permite otorgar un adecuado asesoramiento genético a los padres en cuanto al pronóstico del paciente y futuro reproductivo de la pareja.
MATERIAL Y MÉTODOS
Se realizó un estudio descriptivo de tipo retrospectivo de pacientes con presencia de defectos al nacimiento, en el Instituto Nacional de Perinatología en el periodo comprendido de enero a diciembre del 2003, valorados por el Departamento de Genética. Se incluyeron a los pacientes con alteraciones cromosómicas numéricas y estructurales, detectadas por estudio citogenético realizado en linfocitos de sangre periférica, líquido amniótico o sangre de cordón umbilical por medio de bandas GTG, con una resolución mínima de 450 bandas.
En el INPer durante el periodo arriba mencionado, nacieron 5,795 pacientes de los cuales 5,650 fueron recién nacidos y 145 óbitos. El Departamento de Genética revisó a 330 neonatos por sospecha de alteraciones genéticas y/o congénitas, de los cuales 189 pacientes (52.7%) presentaron defectos al nacimiento. El resto de los recién nacidos fue valorado por diferentes motivos como: antecedente familiar de defecto congénito y/o enfermedad genética, patología materna y/o paterna, exposición a teratógenos y sospecha de enfermedad genética.
Las indicaciones para realizar estudio citogenético prenatal, fueron: edad materna avanzada, antecedente de madre portadora de translocación balanceada y defectos estructurales por ultrasonografía (USG) de alta definición. Las indicaciones para realizar cariotipo en linfocitos de sangre periférica al nacimiento, fueron la presencia de múltiples defectos congénitos y la sospecha de alteración cromosómica.
A los pacientes con alteración cromosómica estructural se les realizó hibridación in situ con fluorescencia (FISH, por sus siglas en inglés) con el objeto de completar y/o corroborar el diagnóstico citogenético en los casos pertinentes. Se excluyeron los recién nacidos con defectos congénitos con cariotipo normal.
RESULTADOS
Durante el año 2003, 3.26% (189/5,795) de los pacientes nacidos en el INPer presentaron defectos al nacimiento, de los cuales veintisiete pacientes 27/189 (14.2%) mostraron un cariotipo anormal: 21 (77.7%) presentaron alteraciones cromosómicas numéricas y en seis (22.2%) se encontró una alteración cromosómica estructural (Tabla 1). De manera que las alteraciones cromosómicas y estructurales representaron 0.46% (27/1,595) de los pacientes nacidos en el INPer.
El promedio de la edad materna de los pacientes con alteración numérica fue de 34.3 años con un intervalo de 18 a 46 años y se encontró que 52% de las madres tenían una edad mayor o igual a 35 años (Tabla 2). En nuestros pacientes 21 estudios citogenéticos fueron realizados al nacimiento por la sospecha clínica del Departamento de Neonatología y Genética; mientras que en seis casos los diagnósticos fueron realizados prenatalmente por alteraciones estructurales en el ultrasonido nivel II.
Aspectos clínicos de los pacientes con alteraciones numéricas
Veintiún pacientes presentaron alteraciones cromosómicas numéricas; en cuatro casos el diagnóstico se realizó en etapa prenatal por amniocentesis y cordocentesis (Tabla 3). La trisomía 21 regular fue el diagnóstico más frecuente, encontrado en 18 pacientes con diagnóstico clínico de síndrome de Down al nacimiento, caracterizado por la presencia de múltiples defectos menores, cardiopatía y en algunos casos atresia intestinal. Diecisiete pacientes presentaron una trisomía regular con 47 cromosomas en todas las células analizadas y un paciente presentó trisomía 21, secundaria a un rearreglo estructural. Los 17 pacientes fueron referidos a instituciones especializadas para el tratamiento de pacientes con síndrome de Down. A continuación se comentan, algunos de los casos más representativos de este grupo de alteraciones.
Una paciente del sexo femenino con hy–drops fetal presentó monosomía del X, el diagnóstico fue realizado en forma prenatal por medio de cordocentesis: la paciente presentó muerte fetal.
Otra paciente del sexo femenino presentó múltiples anomalías por USG de alta definición, realizándose amniocentesis con resultado 47,XX + 18, en 15 células analizadas de líquido amniótico proveniente de cuatro cultivos primarios. A la revisión clínica se encontró microtia tipo I bilateral, retromicrognatia, fascies pequeñas, manos con sobreposición de dedos, pliegues aberrantes. Pie equinovaro bilateral y talones prominentes: la paciente presentó muerte neonatal tardía.
Un paciente del sexo masculino fue valorado al nacimiento por la presencia de microcefalia criptorquidia bilateral, defecto lumbosacro, polidactilia postaxial en manos y pies con sobreposición de dedos. El resultado de cariotipo en linfocitos correspondió a 47,XY + 13, en 15 células analizadas: el paciente presentó muerte neonatal temprana.
Otro paciente más, fue valorado por dismorfia facial con puente nasal ancho y deprimido, pabellones auriculares laxos, corazón con comunicación interventricular posterior, genitales externos masculinos, hoyuelo sacro, manos y pies con pliegues profundos y aumento de tejido subcutáneo, ausencia de uña del quinto dedo de mano derecha y del cuarto y quinto ortejos del pie izquierdo y quinto ortejo del pie derecho. La radiografía de tórax mostró 13 costillas bilaterales con alteraciones en la forma. El ultrasonido transfontanelar reportó agenesia de cuerpo calloso y colpocefalia. El resultado de cariotipo se reportó como 46,XY(10)/ 47,XY + 8(10), en 20 células analizadas de sangre periférica (Figura 2). El paciente fue referido a una institución pediátrica de tercer nivel.

Aspectos clínicos de los pacientes con alteraciones cromosómicas estructurales (Tabla 3)
Seis pacientes presentaron alteraciones cromosómicas estructurales. En dos casos, estas alteraciones eran balanceadas y en cuatro pacientes desbalanceadas. Las características principales de los cuadros clínicos se presentan a continuación:
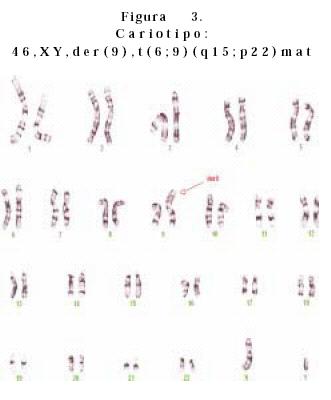
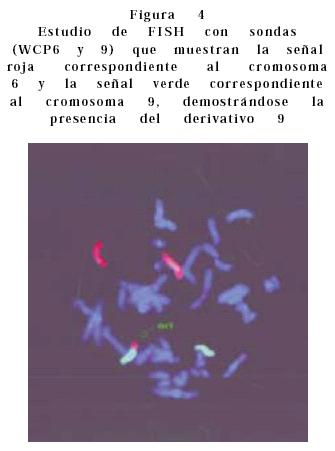
• Caso 1. Se valoró a recién nacida por la presencia de hipertelorismo ocular con puente nasal ancho, boca con labio y paladar hendido bilateral, pabellones auriculares de baja implantación, cuello corto, tórax ancho, corazón con insuficiencia bivalvular severa, manos con camptodactilia, edema distal y polidactilia postaxial de mano derecha. Pies con sobreposición de dedos y edema bilateral. El USG renal reportó múltiples quistes renales bilaterales. El resultado en 15 células de linfocitos de sangre periférica reportó un cariotipo 46, XY, der (9). Luego de lo cual se realizó cariotipo a los padres reportando los siguientes resultados: madre, 46, XX, t(6;9) (q15;p22); padre 46, XY. El cariotipo del paciente fue 47, XY der (9)t(6;9)(q15;p22)mat. (Figura 3). Se realizó estudio de citogenética molecular por medio de la técnica de FISH, el cual confirmó el diagnóstico anterior (Figura 4). La paciente presentó muerte neonatal tardía.
• Caso 2. Se valoró a paciente masculino con reporte de ultrasonido nivel II, con hydrops fetal, retraso en el crecimiento intrauterino asimétrico, fosa posterior con escotadura, arteria umbilical única, columna vertebral con hemivértebras y alteración en la posición de ambos pies. Se realizó amniocentesis, reportándose un resultado de 46, XY,–13 + mar. Se realizó estudio de FISH con sonda centromérica del cromosoma 13 y se encontraron dos señales para dicho cromosoma, con lo que se confirmó que el marcador observado provenía del cromosoma 13; con resultado 46, XY, r(13) (p11;q14). Al nacimiento presentó dismorfia facial, frontal amplio, hipertelorismo ocular, opacidad en ojo derecho, pabellón auricular izquierdo con apéndices preauriculares y ambos pabellones de baja implantación. Cuello corto con piel redundante, corazón con probable comunicación aortopulmonar, genitales externos ambiguos (caracterizados por micro–pene con hipospadias coronal), escroto "en chai" con criptorquidia bilateral, ano imperforado, manos con oligodactilia, uñas convexas. Pie equinovaro bilateral, pie derecho con cuatro ortejos e izquierdo con sindactilia cutánea entre primero y segundo ortejos. En la radiografía se encontró alteración en la segmentación costovertebral, con 11 arcos costales del lado derecho y 13 del lado izquierdo, hemivértebras torácicas. El paciente presentó muerte neonatal temprana. Se ofreció estudio citogenético a los padres, el cual no se realizó.
• Caso 3. RN masculino gesta 5, de madre de 28 años de edad, portadora de translocación balanceada 46,XX,t(7; 10) (q32;q22), diagnosticada por pérdida gestacional recurrente. A la exploración física no presentó defectos estructurales externos, fenotipo normal, se le proporcionó seguimiento y vigilancia. El resultado del cariotipo en linfocitos de sangre periférica, mostró un resultado 46,XY,t(7; 10) (q32;q22), heredando de la madre la misma translocación por medio de una segregación alterna.
• Caso 4. RN masculino gesta 5 de padres con edad avanzada, motivo por el cual se realiza amniocentesis, la que reporta como resultado una translocación 46,XY,t(7;8) (q31.2;q24.1), aparentemente balanceada. Se realizó estudio citogenético a los padres y se obtuvo un resultado normal en ambos. El USG nivel II reportó masa hiperecogénica, no vascularizada en rodilla izquierda. A la revisión clínica se corrobora el hallazgo prenatal, encontrándose extremidad inferior izquierda con masa de aproximadamente 6x6x4 cm con área de necrosis y laceración en la porción superior, con múltiples vasos pequeños, bien limitada, que afecta la movilidad de la articulación. Reflejos distales presentes en forma bilateral. El paciente fue referido a otra institución especializada para su tratamiento.
• Caso 5. RN del sexo femenino, gesta 3 de padres jóvenes, aparentemente sanos. Hija previa con diagnóstico de insuficiencia tricuspídea. Se realiza USG nivel II, en donde se informa la presencia de una probable cardiopatía con estenosis de la válvula pulmonar, dilatación de la aurícula derecha y ventrículo derecho hipertrófico. A la exploración física presenta cráneo normo–céfalo con fontanela anterior amplia que comunica con sutura metópica y con fontanela posterior. Cara con región frontal amplia, epicanto bilateral, nariz pequeña con puente nasal ancho y plano, narinas hipoplásicas, columnela corta, filtrum largo, paladar íntegro, pabellones auriculares de baja implantación con rotación posterior. Cuello corto, tórax ancho con teletelia, cardiopatía caracterizada por estenosis pulmonar, genitales externos femeninos, extremidades superiores con camptodactilia y desviación cubital del segundo al quinto dedo. Pies con talón prominente, hiperflexión de tobillos. El USG transfontanelar reportó disgenesia de cuerpo calloso. Se realizó estudio citogenético en linfocitos de sangre periférica con resultado 46,XX,der (7)t(2;7)(q36;p11.2), razón por la cual se realiza cariotipo a ambos padres y a la hermana. Los tres resultados se reportaron como normales. La paciente fue referida a otra institución para su tratamiento.
• Caso 6. RN masculino, gesta 4, de madre de 40 años de edad y padre de 41 años, aparentemente sanos. A la exploración física presentó fenotipo compatible con trisomía 21, por lo que se indica estudio citogenético, el cual se reporta 46,XY,dic(21;21) + 21. Se realiza cariotipo a los padres reportándose como normal. El paciente fue referido a otra institución para su seguimiento.
DISCUSIÓN
Las alteraciones cromosómicas numéricas y estructurales se presentaron en 0.46% de los pacientes nacidos en el INPer, lo que está de acuerdo con la literatura reportada5 y comprende 14.2% de los pacientes con defectos congénitos durante este periodo de tiempo. La gran mayoría de las alteraciones numéricas corresponde a la trisomía 21, la cual es causada por un error en la meiosis materna en aproximadamente 90% de los casos. Los errores en la meiosis I se han asociado con un número reducido de quiasmas entre las cromátides del bivalente 21.14 En 17 pacientes con síndrome de Down y en los pacientes con trisomía 13 y 18, los cariotipos correspondieron a una trisomía regular, lo cual se presenta en 95% de los pacientes con síndrome de Down y concuerda con lo reportado en la literatura.15 La asociación entre edad materna avanzada y aneuploidia fue reconocida hace más de 60 años y han sido propuestos diferentes modelos para explicar esta asociación: como la falta de un número y localización adecuada de los quiasmas y/o la disminución en el proceso de recombinación de los ovocitos.16 El asesoramiento genético está basado en la historia familiar, cariotipo del afectado y la edad materna.
En un paciente se encontró un rearreglo muy raro, críptico entre dos cromosomas 21, los cuales se observaron fusionados por los telómeros de los brazos largos. El cromosoma tenía dos centrómeros, uno de los cuales se encontraba inactivado funcionalmente. En general, estos cromosomas son resultado de eventos esporádicos, posiblemente por una translocación entre cromátides hermanas.17 El resultado de los padres fue normal. La madre de este paciente tenía edad materna avanzada, por lo que se puede suponer que inicialmente se originó de una no disyunción en la meiosis materna, seguida por la fusión de los brazos largos de dos cromosomas 21. El fenotipo de este paciente fue similar al de los pacientes con trisomía 21 regular.
La monosomía del cromosoma X se origina por falta de uno de los cromosomas sexuales del padre en 80% de los casos.16 En la mayoría de los casos el error se presenta en la meiosis masculina y probablemente refleja la ausencia de apareamiento entre el bivalente XY, con un error posterior en la disyunción. De los casos con cariotipo 45,X, 95% son letales de forma prenatal. El riesgo de recurrencia para los padres es bajo.
La trisomía del cromosoma 8 en mosaico es viable, la cual se produce por una alterada segregación–no disyunción de los cromosomas homólogos o por un retraso en la anafase de alguno de los homólogos durante el periodo poscigótico. El mosaico de trisomía 8 se presenta en uno de cada 25,000 recién nacidos, con la presencia de defectos múltiples al nacimiento.18 El riesgo de recurrencia para los padres es bajo.
Se encontraron cuatro pacientes con alteración cromosómica desbalanceada, uno de ellos referido anteriormente, que corresponde al paciente con síndrome de Down (46,XY, dic (21;21) +21).
Los tres pacientes restantes, con rearreglos cromosómicos, mostraron al nacimiento múltiples defectos mayores y menores secundarios, probablemente a un desbalance en la dosis génica o a un efecto de posición del rearreglo.
La paciente con el cariotipo 46,XY,der(9) t(6;9)mat, presentó además reversión sexual, ya que sus genitales externos eran femeninos. Esta alteración es secundaria a un trastorno en la diferenciación sexual, ocasionada probablemente por la monosomía parcial del cromosoma 9, en donde se han propuesto dos genes que intervienen en este proceso. La madre de esta paciente presentó una translocación balanceada entre los cromosomas 6 y 9, con un cariotipo 46,XX,t(6;9) (ql 5;p22). Durante la gametogénesis ocurrió una segregación adyacente tipo 1, por lo que la paciente presentó una trisomía parcial del brazo largo del cromosoma 6 y una monosomía parcial del brazo corto del cromosoma 9. Este tipo de segregación es el modelo más frecuente de una segregación anormal en los hijos de portadores balanceados.
La paciente con cariotipo 46,XX,der(7) t(2;7) (q36;pl 1.2) presentó múltiples defectos; sin embargo, los padres de esta paciente presentaron un cariotipo normal, por lo que el riesgo de recurrencia es bajo. Ambas pacientes presentaron muerte neonatal.
El paciente con anillo del cromosoma 13, con cariotipo 46,XYr(13), presentó hydrops de forma prenatal. El modo de formación de un anillo cromosómico es la ruptura en ambos brazos del cromosoma con fusión de los puntos de ruptura y pérdida de los fragmentos distales. Representan una forma de deleción terminal con la característica adicional de ser mitótica mente inestables, debido a un problema mecánico durante la replicación. El fenotipo clásico incluye defectos mayores y retraso mental.19 De los anillos, 99% se presentan de forma esporádica con bajo riesgo de recurren cia para los padres con cariotipo normal.
Un paciente presentó una alteración cromosómica estructural balanceada con cariotipo 46,XY, t(7;10) (q32;q22), heredada por medio de una segregación alterna de la madre. A la exploración física el paciente no presentó ningún tipo de defecto. Tradicionalmente, al encontrarse el mismo cariotipo balanceado en el hijo, no existe un riesgo incrementado de anomalía fenotípica. Sin embargo, Fryns20 reportó un riesgo de 6.4% de defectos estructurales y retraso mental en hijos portadores de la misma translocación de alguno de los padres. Los mecanismos posibles incluyen un defecto no balanceado muy pequeño (críptico), no visible por medio de un cariotipo convencional, la pérdida poscigótica de un cromosoma derivativo en una línea celular, efecto de posición y disomía uniparental.
El paciente con cariotipo 46,XY, t(7;8) (q31.2;q24.1) presentó una masa tumoral en rodilla; el cariotipo de ambos padres fue normal. En apariencia, la translocación se observó como balanceada, por lo que no se puede definir si la tumoración es consecuencia del rearreglo o se presentó en forma coincidental. Cuando una anomalía cromosómica ocurre como un evento nuevo, el riesgo para una enfermedad genética o un defecto estructural se encuentra incrementado. Esto se debe a que pueden existir deleciones o duplicaciones submicroscópicas en el sitio de la ruptura o a cambios funcionales en los genes cerca del punto de ruptura. Warburton,21 informó acerca de un riesgo de 6.1% para algún defecto consénito en caso de una translocación recíproca, un riesgo de 3.7% para una translocación Robertsoniana y 9.4% para una inversión. Algunas translocaciones cambian la posición de protooncogenes alterando el ciclo celular con el desarrollo de tumores o leucemia.22
En los casos de alteración cromosómica desbalanceada se debe realizar el estudio cromosómico familiar para poder confirmar el diagnóstico y otorgar riesgo de recurrencia.
En 22% de los casos presentados el diagnóstico se realizó en la etapa prenatal, este porcentaje es bajo debido a que las pacientes son derivadas a nuestra institución en forma tardía.
Se puede concluir que la mayoría de las alteraciones cromosómicas numéricas y estructurales se presentan con múltiples defectos al nacimiento, lo que puede ocasionar la muerte fetal, muerte neonatal o durante la infancia.
Por su parte, los pacientes con trisomía 21 tienen una sobrevida de 60 años en países desarrollados, por lo que es importante que la estimulación temprana se realice a partir de los 40 días de vida. La sospecha de este tipo de alteraciones cromosómicas se debe realizar en pacientes con múltiples defectos mayores y es importante que se realicen los estudios necesarios a la familia para poder brindar un adecuado asesoramiento genético.
Finalmente, es importante señalar que las técnicas de citogenética molecular aplicadas al diagnóstico preimplantación permiten a los padres portadores de rearreglos balanceados tener hijos sanos.
REFERENCIAS
1. Ling EW, Sosuan LC, Hall JC. Congenital anomalies: an increasingly important cause of mortality and workload in a neonatal intensive care unit. Am J Perinat 1991; 8: 164–9. [ Links ]
2. Aase JM. Diagnostic Dysmorphology. 2a ed. Nuevo México, USA: Plenum Medical Book Company. 1990, p. 7. [ Links ]
3. Lejeune J, Gautier M, Turpin R. Etude des chromosomes somatiques de neuf enfants mongoliens. CR Acad Sci 1959; 248: 1721–2. [ Links ]
4. Ford CE, Jones KW, Polani PE, De Almeida JC, Briggs JH. A sex chromosomal anomaly in a case of gonadal dysgenesis (Turner's syndrome). Lancet 1959; 1: 711–3. [ Links ]
5. Borgaonkar DS. Chromosomal variation in man. A catalog of chromosomal variants and anomalies, 7a ed. New York: Wiley–Liss; 1994. [ Links ]
6. Hassold T, Abruzzo M, Adkins K, Griffin D, Merrill M, Millie E, Saker D, Shen J, Zaragoza M. Human aneuploidy: incidence, origin and etiology. Environ Mol Mutagen 1996; 28: 167–75. [ Links ]
7. Gardner RJ, Sutherland GR. Chromosome abnormalities and genetic counseling. 3a ed. Australia; Oxford; 2004, p. 334–62. [ Links ]
8. Simpson JL, Elias S. Genetics in obstetrics and gynecology. 3a ed. Texas, USA: Saunders; 2003. [ Links ]
9. Jacobs PA. Structural rearrangements of the chromosomes in man. En: Hook EB, Porter IH (eds.). Population cytogenetics. studies in humans. New York: Academic Press; 1977. p. 81–97. [ Links ]
10. Epstein CJ. Mechanisms of the effects of aneuploidy in mammals. Ann Rev Genet 1988; 22: 51–75. [ Links ]
11. Shapiro BL. Down syndrome —a disruption of homeostasis. Am J Med Genet 1983; 14: 241–69. [ Links ]
12. Andreff M, Pinkel D. Introduction to fluorescence in situ hybridization. Principles and clinical applications, la ed. Nueva York, USA. p. 3–10. [ Links ]
13.Abramsky L, Chappie J. Prenatal diagnosis. The human side. London: Chapman and Hall; 1994. [ Links ]
14. Shaffer L, Lupski J. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet 2000; 34: 297–329. [ Links ]
15. Schinzel A. Human cytogenetics database. Oxford: 2a ed. Oxford University Press; 1994. [ Links ]
16. Hassold T, Arnovitz K, Jacobs PA, May K, Robinson D. The parental origin of the missing or additional chromosome in 45,X and 47, XXX females. Birth Defects 1991; 26: 297–304. [ Links ]
17. Pángalos C, Théophile D, Sinet PM, Marks A, Stamboulieh–Abazis D, Chettouh Z, et al. No significant effect of monosomy for distal 21q22.3 on the Down syndrome phenotype in "mirror" duplications of chromosome 21. Am J Hum Genet 1992; 51: 1240–50. [ Links ]
18. Nielsen J, Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13–year incidence study in Arhus. Denmark. Hum Genet 1991; 87: 81–3. [ Links ]
19. Lorentz CP, Jalal SM, Thompson DM, Babovic–Vuksanovic D. Mosaic r(13) resulting in large deletion of chromosome 13q in a newborn female with multiple congenital anomalies. Am J Med Genet 2002; 111: 61–7. [ Links ]
20. Fryns JP, Kleczkowska A, Kubién E, Van der Berghe H. On the excess of mental retardation and/or congenital malformations in apparently balanced reciprocal translocations. A critical review of the Leuven data 1966–1991. Genet Counsel 1992; 2: 185–94. [ Links ]
21.Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet 1991; 49: 995–1013. [ Links ]
22. Rowley JD. The role of chromosome translocations in levkemogenosis. Semin Hematol 1999; 36: 559–72. [ Links ]

