










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArchivos de neurociencias (México, D.F.)
versão On-line ISSN 1028-5938versão impressa ISSN 0187-4705
Arch. Neurocien. (Mex., D.F.) vol.10 no.3 Ciudad de México Jul. 2005
Artículo original
Oligodendrogliomas en el Instituto Nacional de Neurología y Neurocirugía: comportamiento biológico en una población definida
Oligodebdrogliomas the National Institute of Neurology and neurosurgery mvs: biologic behavior in a defined population
Sergio Moreno–Jiménez1, Mario Alonso–Vanegas1, Antonio Bramasco Aviléz1, Cuauhtémoc García–Pastor1, Javier Terrazo–Lluch1, Martha Tena3, Lucinda Aguirre2
1 Instituto Nacional de Neurología y Neurocirugía. Departamento de Neurocirugía
2 Instituto Nacional de Neurología y Neurocirugía. Departamento de Neuroinmunología
3Instituto Nacional de Neurología y Neurocirugía. Departamento de Patología
Correspondencia:
Sergio Moreno–Jiménez.
Instituto Nacional de Neurología y Neurocirugía.
Insurgentes Sur # 3877 Col. La Fama.
14269 México D.F.
E–Mail: sermorjim@hotmail.com
Recibido: 25 febrero 2005.
Aceptado: 11 marzo 2005.
RESUMEN
Los oligodendrogliomas corresponden entre el 2 y 5% de los tumores intracranerales siendo diagnosticados en la 4º ó 5º décadas de la vida. La localización más frecuente es el lóbulo frontal. La cirugía es la piedra angular del tratamiento utilizado. Material y métodos: entre enero de 1990 y enero del 2000, 28 pacientes tuvieron diagnóstico de oligodendroglioma. Se revisaron los expedientes en busca de localización, signos y síntomas de presentación, diagnóstico preoperatorio, estirpe histológica, tipo de tratamiento recibido y complicaciones. Resultados: el sitio de localización más frecuente fue el lóbulo frontal (43%), mientras que el síntoma de presentación principal fue epilepsia (61%). El diagnóstico preoperatorio fue glioma de bajo grado en 39% de los casos. Se realizó resección parcial en 36% de los casos. La cirugía fue realizada por un neurocirujano recibido en la mitad de los casos y por un residente de último año en la otra mitad. Recibió radioterapia fraccionada el 71% de los pacientes, mientras que 21% recibieron quimioterapia. El diagnóstico histopatológico fue oligodendroglioma en 71% de los casos y oligodendroglioma anaplásico en el 29%. La mortalidad operatoria fue del 8%. Conclusiones: los resultados obtenidos en el estudio son similares a los descritos en la literatura.
Palabras clave: gliomas, oligodendroglioma, oligodendroglioma anaplásico, cirugía.
ABSTRACT
Oligodendrogliomas represent 2 to 5% of the intracraneal tumors. Usually these tumors are diagnosed between the 4th and the 5th decades of life. The most commonly site is the frontal lobe. Surgery is the milestone of the treatment approach.
Material and methods: between January 1990 and January 2000, 28 patients had a diagnosis of oligodendroglioma. We looked for localization, signs and symtoms of presentation, peroperative diagnosis, histopathology, type of treatment received and complications.
Results: the most frecuent site was the frontal lobe (43%), while the main symptom of presentation were seizures (61%). The preoperative diagnosis with clinical data and imaging was low–grade glioma in 39% of the cases. Partial resection was performed in 36% of the cases being the most prevalent of all treatments. The surgery was performed by neurosurgeons in half the cases and by residents of the last year in the other half. Seventy one porcent of patients were treated with radiotherapy, and 21% with chemotherapy. The hystopathologic diagnosis was oligodendroglioma in 71% and anaplastic oligodendroglioma in 29%. The operative mortality was 8%. Conclusion: our results are similar to the ones described
Keywords: gliomas, oligodendroglioma, anaplastic oligodendroglioma, surgery.
El propósito del trabajo es describir el comportamiento biológico del oligodendroglioma en la población del Instituto Nacional de Neurología y Neurocirugía en un periodo definido.
Los oligodendrogliomas corresponden entre el 2 y 5% de los tumores intracraneales. Por lo general son diagnosticados en la 4º ó 5º décadas de la vida, con un discreto predominio en los hombres. Estos tumores son típicamente vascularizados y con frecuencia calcificados. La localización más común es el lóbulo frontal, aunque se pueden encontrar en cualquier lugar del sistema nervioso central1. La cirugía ha sido la piedra angular del tratamiento, y se utilizan la quimioterapia y la radioterapia como tratamientos adyuvantes2.
Epidemiología
Los oligodendrogliomas son tumores raros del sistema nervioso central (SNC). Corresponden entre el 4 y 7% de todos los tumores primarios del SNC1,3,8. Un estudio Noruego reportó 208 oligodendrogliomas confirmados en una población de 6,180 tumores intracraneales primarios, representando un 4.2%. Esto da una incidencia de 240 casos nuevos por año en esa población2. Algunos estudios que incluyen tanto oligodendrogliomas como gliomas mixtos con contribución de estirpe oligodendroglial reportan que corresponden entre un 5 y un 10% de todos los gliomas. Los tumores de bajo grado tienden a ocurrir en pacientes más jóvenes6. Por lo general son diagnosticados en la 4º ó 5º décadas de la vida, y hay un ligero predominio en el género masculino6,7. La frecuencia de este tumor en pacientes pediátricos (menos de 20 años) es mucho menor que en adultos, representando únicamente entre el 1 y el 2% de todas las neoplasias primarias del SNC9. Los reportes de casos de oligodendrogliomas en niños han sido esporádicos10–16.
Cuadro clínico
El cuadro clínico con el que se presentan los pacientes con oligodendrogliomas depende en gran parte de la localización del tumor. MorK et al, reportaron 208 casos de oligodendrogliomas de los cuales todos fueron supratentoriales. La mitad afectó al lóbulo frontal, una tercera parte al lóbulo parietal, una cuarta parte al temporal y una decimosexta parte al occipital. Solamente hubo seis lesiones predominantemente intraventriculares. El sitio más frecuente fue la sustancia blanca del lóbulo frontal (111 casos, 53%)2. En 11 pacientes menores de 20 años de edad se encontraron cuatro tumores en el lóbulo frontal, cuatro en el parietal, dos en el temporal y uno en el occipital9. La epilepsia es el síntoma inicial más común tanto en la población adulta como en la pediátrica, representando 57 y 64% respectivamente. Después se refieren cefalea (22%), alteración de funciones mentales (10%) y vértigo/náusea (9%). En los niños el segundo lugar lo ocupan la náusea y el vómito (55%)2,9.
Diagnóstico por imagen
En radiografías simples de cráneo, cerca del 47% de todos los oligodendrogliomas presentan alguna calcificación17. La frecuencia de calcificaciones ha sido reportada tan alta como 91 % en detecciones por tomografía18, y es un hallazgo casi universal en el estudio histológico. A medida que se ha logrado diagnosticar a los pacientes con oligodendrogliomas de una manera más rápida, la tendencia es a encontrar cada vez menos calcificaciones. En la tomografía no contrastada los oligodendrogliomas se ven comúnmente en forma heterogénea, como una mezcla de áreas hipodensas e isodensas, calcificaciones y, en ocasiones hemorragias. El efecto de masa es por lo general proporcional al tamaño del tumor aunque puede ser menor a lo esperado. El edema perilesional puede ser de leve a moderado19. Otra característica que distingue a los oligodendrogliomas de otros tumores es la presencia de erosión festoneada de la tabla interna del cráneo. En la imagen de resonancia magnética el oligodendroglioma se presenta como una masa que puede ser heterogénea en todas las secuencias20. Se pueden observar hiperintensidades en sitios de hemorragias previas en secuencia T1WI. Puede ser necesario utilizar una técnica con echogradiente para identificar pequeñas calcificaciones. Otra técnica utilizada más en la actualidad para diferenciar principalmente entre una recidiva tumoral o radionecrosis en pacientes ya tratados, es la resonancia magnética con espectroscopia, encontrando un aumento en los picos de colina y creatina en relación al de N–acetil–aspartato21, además de la tomografía por emisión de positrones.
Patología
Los oligodendrogliomas se caracterizan por tener células con núcleos redondos y ovales, cuyo nucléolo es ligeramente prominente. Las prolongaciones citoplásmicas de los tumores bien diferenciados son muy escasas y cortas. Los halos perinucleares le dan el aspecto de "huevo estrellado", y se considera un artificio de fijación (figura 1). En las formas anaplásicas o malignas, los oligodendrogliomas, al igual que otros gliomas, presentan notable pleomorfismo, anaplasia, necrosis y vasos tumorales con gran prominencia de las células endoteliales.

Tratamiento
En particular, los oligodendrogliomas son difíciles de resecar por completo por su carácter infiltrativo; sin embargo, la cirugía resectiva continúa siendo la piedra angular del tratamiento22. Un meta–análisis reciente confirmó que existe un aumento en la sobrevida en pacientes que son tratados con cirugía y radioterapia en comparación con aquellos que sólo se operan23. Se reportaron las diferentes modalidades de tratamiento en 54 pacientes. Los datos de seguimiento para el análisis de sobrevida a 5 años se obtuvieron en 35 de estos pacientes. Once pacientes fueron solamente operados y 24 recibieron radioterapia posoperatoria. El índice de sobrevida a 5 años para el grupo quirúrgico fue de 82%, mientras que para el grupo de cirugía y radioterapia fue de 100%. La dosis varió de 5,300 a 7,000 cGy dada en un tiempo total de 49 a 66 días6. Para algunos autores, la quimioterapia es la principal modalidad de tratamiento adyuvante posterior a la resección quirúrgica, y la radioterapia se reserva para la transformación anaplásica si es que ocurre24,28. La mayoría de los oligodendrogliomas responden a la quimioterapia, por lo general en menos de 3 meses, frecuentemente con una reducción en el tamaño. La respuesta es variable en su grado y duración25. La pérdida de un alelo en el cromosoma 1p, y la pérdida combinada de los brazos cromosómicos 1p y 19q, están asociados con la respuesta a la quimioterapia y con una mayor sobrevida libre de tumor después del tratamiento con quimioterapia26,27.
Se ha usado la combinación de procarbazina, lomustina y vincristina (PCV)28, o con procarbazina, vincristina, CCNU o 6–tioguanina.
Genética
Se ha reportado el gen P18INK4C como el gen supresor tumoral involucrado en los oligodendrogliomas en 1p32. En ese estudio se encontró una mutación de p18 en ciertos pacientes en los cuales había recurrencia tumoral en forma de oligodendroglioma anaplásico, pero no en los tumores primarios de bajo grado. De esta manera, se concluyó que las alteraciones del p18 están involucradas en la progresión tumoral en un subgrupo de oligodendrogliomas29.
Pronóstico
Los oligodendrogliomas puros tienen un mejor pronóstico que los tumores mixtos (oligoastrocitomas) y, estos, con mejor pronóstico que los astrocitomas. Un componente oligodendroglial, fuera de la cantidad, confiere un mejor pronóstico. Se ha reportado una sobrevida a 10 años de 10 a 30% para pacientes con tumores sólo o predominantemente de estirpe oligodendroglial30. La presencia de calcificaciones se asocia a un mejor pronóstico2,31.
MATERIAL Y MÉTODOS
El Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez cubre gran parte de la población de la Ciudad de México y provincia. En el periodo comprendido entre 1990 y 2000, fueron atendidos un total de 28 pacientes con diagnóstico histopatológico de oligodendroglioma u oligodendroglioma anaplásico.
Evaluación histológica
Los cortes histológicos fueron evaluados por un patólogo con conocimiento de los datos clínicos. Se utilizó la definición de oligodendroglioma de la clasificación de la Organización Mundial de la Salud (WHO por sus siglas en inglés), es decir: un tumor compuesto predominantemente por células oligodendrogliales. Las células con halos perinucleares fueron consideradas como oligodendrogliales. Cuando se encontraba más de un 25% de células neoplásicas astrocitarias, entonces se clasificaron como tumores mixtos (oligoastrocitomas) y fueron sacados del estudio32.
Tratamiento
El tratamiento consistió en cirugía con resección parcial, resección total, biopsia, lobectomía temporal o lobectomía frontal, además de radioterapia y quimioterapia en algunos casos.
Revisión del seguimiento
La muerte operatoria se definió como la que se presentaba dentro del primer mes después de la primera cirugía, fuera de la causa. El rango de seguimiento fue de 0.3 a 11.4 años con una media de 8.2 años.
Análisis estadístico
Se revisaron los expedientes de los 28 pacientes con respecto a la edad al momento del diagnóstico, género, lateralidad manual, síntomas de presentación, meses de evolución antes del diagnóstico, localización, cirugía realizada, tratamiento adyuvante aplicado, tiempo de seguimiento y complicaciones. Todas las variables fueron evaluadas individualmente con frecuencias simples, y con medidas de tendencia central y dispersión cuando aplicaba.
RESULTADOS
La edad de los 28 pacientes tuvo una media de 44 años (18–73).
Epidemiología
De 2,565 pacientes operados en el Instituto Nacional de Neurología y Neurocirugía con diagnóstico de tumores de origen neuroepitelial de 1964 hasta el 2003, solamente 99 correspondieron a diagnóstico de oligodendroglioma. Esto corresponde al 3.9% del total. En el periodo de estudio de 1990 al 2000 la prevalencia fue del 4.2%.
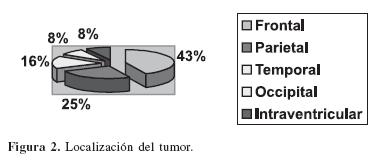
Localización
Todos los oligodendrogliomas fueron supratentoriales. La localización fue el lóbulo frontal 43%, seguido por lóbulo parietal 25%, lóbulo temporal 16% y, por último, lóbulo occipital 8% e intraventricular 8%. Algunos tumores comprometían más de un solo lóbulo por lo que la suma total nos da más de 28 (figura 2).
Signos y síntomas
El síntoma más frecuente fue la presencia de crisis convulsivas que se presentaron en 17 pacientes (61%). De los 17 pacientes con epilepsia, ocho pacientes presentaron crisis convulsivas tónico clónico generalizadas de inicio (40%), crisis parciales simples en siete pacientes (35%), y crisis parciales complejas en cinco pacientes (25%) (tabla 1). Quince pacientes presentaron cefalea (54%), cinco (18%) afección motora, y un paciente (4%) afección sensitiva. Cinco pacientes (18%) presentaron disfasia, cuatro de tipo motor y uno de tipo mixta. Ocho pacientes se presentaron con alteración del estado de alerta (29%) no relacionado a estado postictal, seis con somnolencia y dos con estupor superficial. Un paciente (4%) presentó parálisis facial central y uno más (4%) presentó fiebre. Once pacientes (39%) presentaron papiledema al momento del primer examen neurológico. Estos once pacientes presentaron también cefalea, náusea y vómito, diagnosticándose hipertensión intracraneana (tabla 2). La suma de los porcentajes es mayor a 100 debido a que un solo paciente podía presentar varios síntomas.


Diagnóstico preoperatorio
Los diagnósticos preoperatorios con la clínica y los estudios de imagen, dados por parte de los residentes de neurocirugía fueron los siguientes. Once (39%) como gliomas de bajo grado, cuatro (14%) como oligodendrogliomas u oligoastrocitomas, siete (25%) como astrocitomas de alto grado, tres (11%) como metástasis, dos (7%) como meningiomas, y uno (4%) como neurocisticercosis, malformación arteriovenosa, papiloma de plexos coroides y toxoplasmosis (tabla 3). El porcentaje no suma 100 ya que en varios casos se dio más de un diagnóstico presuntivo.
Resultado de patología
El reporte de histopatología fue de oligodendroglioma en 20 pacientes (71%) y de oligodendroglioma anaplásico en ocho (29%).
Tipo de tratamiento
El tratamiento fue quirúrgico en todos los pacientes. En 10 pacientes se realizó una resección parcial (36%), en tres (11%) una resección total, se hizo biopsia en cinco pacientes (18%). En ocho pacientes se realizó una lobectomía frontal (28%) mientras que en dos (7%) una lobectomía temporal (tabla 4). Catorce pacientes fueron operados por un médico adscrito al servicio de neurocirugía y catorce por un médico residente. Veinte pacientes recibieron tratamiento posoperatorio con radioterapia con dosis entre 5,000 y 6,000 cGy (71%). Solamente seis pacientes recibieron quimioterapia (21%) a base de BCNU, 5FU, carmustina y procarbazina.
Complicaciones
Las complicaciones que se presentaron fueron déficit neurológico focal en ocho pacientes (29%), cuatro pacientes con hematomas posoperatorios del lecho o hemorragia intraventricular (14%), fístula posoperatoria de líquido cefalorraquídeo en dos pacientes (7%), epilepsia posoperatoria en un paciente (4%), y neuroinfección en otro (4%). Tres pacientes presentaron muerte operatoria (11%), uno de ellos por broncoaspiración posterior a una crisis convulsiva (tabla 5)

DISCUSIÓN
Los resultados obtenidos en este trabajo son muy similares a los encontrados en la literatura. La localización más frecuente fue en el lóbulo frontal (43%). Mork reportó la mitad (53%) de los tumores en esta misma localización 2. El síntoma más frecuente fue la epilepsia encontrada en el 61 % de los casos en comparación con 57% de los casos reportados por Earnest. El diagnóstico preoperatorio fue de glioma de bajo grado en 39% de los casos por lo que es indispensable tener un alto índice de sospecha clínica y radiológica. En las series revisadas no se hace mención acerca del diagnóstico preoperatorio.
REFERENCIAS
1. Nijjar TS, Simpson WJ, Gadalla T, Math M, McCartney M. Oligodendroglioma The Princess Margaret Hospital experience (1958–1984). Cancer 1993; 71:4002–6. [ Links ]
2. Mork SJ, Lindegaard KF, Halvorsen TB. Oligodendroglioma: Incidence and biological behavior in a defined population. J Neurosurg 1985; 63:881–9. [ Links ]
3. Shaw EG, Scheithauer BW, O'Fallón JR. Oligodendrogliomas: The Mayo Clinic experience. J Neurosurg 1992; 76:428–34. [ Links ]
4. Lindegaard KF, Mork SJ, Eide GE Statistical analysis of clinicopathological features, radiotherapy, and survival in 170 cases of oligodendroglioma. J Neurosurg 1987; 67:224–30. [ Links ]
5. Russell DS, Rubenstein LJ. Eds. Pathology of tumors of the central nervous system. Baltimore: Williams and Wilkins, 1989. [ Links ]
6. Chin HW, Hazel JJ, KimTH, Webster JH. Oligodendrogliomas. I. A clinical study of cerebral oligodendrogliomas. Cancer 190; 45:1458–66. [ Links ]
7. Earnest FIN, Kernohan JW, Craig WM. Oligodendrogliomas. A review of two hundred cases. Arch Neurol Psychiatry 1950; 63:964–76. [ Links ]
8. Horrax G, Wu WQ. Postoperative survival of patients with intracranial oligodendroglioma with special reference to radical tumor removal. J Neurosurg 1951 ;8:473–9. [ Links ]
9. Dohrmann GJ, Farwell JR, Flannery JT. Oligodendrogliomas in Children. Surg Neurol 1978; 10:21–5. [ Links ]
10. Backus RE, Millichap JG. The seizure as a manifestation of intracranial tumor in childhood. Pediatrics 1962; 29:978–84. [ Links ]
11. Berkheiser SW. Oligodendrogliomas in the young–age group. J Neurosurg 1956; 13:170–5. [ Links ]
12. Farwell JR, Dormán GJ, Flannery JT. Central nervous system tumors in children. Cancer 1977;40: 3123–32. [ Links ]
13. Freeman L, Feigin I. Oligodendroglioma with 35–year survival. J Neurosurg 1963; 20:363–5. [ Links ]
14. Gass A, Van Wagenen WP. Oiigodendrogiioma in pre–adolescence. Report of a case. J Neurosurg 1950; 7:374–6. [ Links ]
15. Roberts M, German WJ. Oiigodendrogiioma: a 40–year survival. Case report. J Neurosurg 1969; 31:355–7. [ Links ]
16. Weir B, Elvidge AR. Oiigodendrogiiomas: an analysis of 63 cases. J Neurosurg 1968; 29:500–5. [ Links ]
17. Kalan C, Burrows EH. Calcification in intracranial gliomata. Br J Radiol 1962; 35:589–602. [ Links ]
18. Vonofakos D, Marcu H, Hacker H. Oiigodendrogiiomas: CT patterns with emphasis on features indicating malignancy. J Comut Assist Tomogr 1979; 3:783–8. [ Links ]
19. Rees J, Lee SH, Smirniotopoulos. Primary brain tumors in adults. In: Lee SH, Rao KCVG, Zimmerman RA, eds. CranialMRIand CT. McGraw–Hill International Edition 1999. [ Links ]
20. Lee YY, Van Tassel PV. Intracranial oligodendrogliomas:lmaging findings in 35 untreated cases. AJNR 1989; 10:119–27. [ Links ]
21. Vigneron DB, Nelson SJ. Magnetic resonance spectroscopy. In: Bernstein M, Berger MS, eds. Neuro–oncology the essentials. Thieme New York; 2000; 99–113. [ Links ]
22. Plunkett SR. Conventional radiotherapy of specific central nervous system tumors. In: Wilkins RH, Rengachary SS, eds. Neurosurgery. McGraw–Hill 1996. [ Links ]
23. Shimizu KT, Tran LM, Mark RJ, Selch MT. Management of oiigodendrogiiomas. Radiology 1993;186:569–72. [ Links ]
24. Fortin D, Cairncross GJ, Hammond RR. Oiigodendrogiioma: An appraisal of recent data pertaining to diagnosis and treatment. Neurosurgery 1999; 45:1279–91. [ Links ]
25. Cairncross JG, McDonald D, Ludwin S Chemotherapy for anapiastic oiigodendrogiioma. J Clin Oncol 1994; 12: 2013–21. [ Links ]
26. Cairncross JG, Ueki K, Zlatescu MC. Specific genetic predictors of chemotherapeutic response and survival in patients with anapiastic oiigodendrogiiomas. J Natl Cancer Inst 1998; 90:1473–9. [ Links ]
27. Ino Y, Zlatescu MC, Sasaki H. Long Survival and therapeutic responses in patients with histologically disparate high–grade gliomas demonstrating chromosome 1p loss. J Neurosurg 2000; 92:983–90. [ Links ]
28. Streffer J, Schabert M, Bamberg M A role for preirradiation PCV chemotherapy for oligodendroglial brain tumors. J Neurol 2000; 247:297–302. [ Links ]
29. He J, Hoang–Xuan, Marie Y. P18 tumor suppressor gene and progression of oiigodendrogiiomas to anaplasia. Neurology 2000; 55:867–9. [ Links ]
30. Gonzales M, Sheline GE. Treatment of oiigodendrogiiomas with or without postoperative radiation. J Neurosurg 1998; 68:684–8. [ Links ]
31. Sun Genka S, Shitara N. Factors Possibly Influencing the Prognosis of Oiigodendrogiioma. Neurosurgery 1988;22:886–91. [ Links ]
32. Félix IA. Tumores de células neurogliales. En: Félix IA ed. Atlas de neuropatología. Auroch 2000; 1–26. [ Links ]

