










Servicios Personalizados
Revista
Articulo
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de neurociencias (México, D.F.)
versión On-line ISSN 1028-5938versión impresa ISSN 0187-4705
Arch. Neurocien. (Mex., D.F.) vol.9 no.4 Ciudad de México dic. 2004
Reporte de caso
Síndrome de Sturge Weber con crisis epilépticas y calcificaciones intracraneales bilaterales en el periodo neonatal
Sturge Weber syndrome with epilepsy and bilateral intracranial calcifications at the neonatal period
A. Fernández Jaén, R. Sánchez Jacobb, E. Ramos Boludac, C. Nevado Jiménez,
S. Simo Segovia, V. González Ojeda, A. Alonso Gómez, L. López Ibor
Sección de Neuropediatría. Servicio de Radiología y Neurorradiología. Servicio de Pediatría. Servicio de Neurología y Neurofisiología.
Correspondencia:
Dr. Fernández Jaén.
Servicio de Neurología Infantil. Hospital La Zarzuela.
C/ Pleyádes 25.
28023 Aravaca. Madrid.
E–mail: ferjaen@nacom.es
RESUMEN
El síndrome de Sturge Weber es un trastorno neurocutáneo infrecuente, caracterizado por la asociación de un angioma venoso de la piamadre con un nevus rojo–vinoso en la cara. La afectación bilateral del nevus no es infrecuente, habiéndose observado en el 30% de los pacientes; sólo el 25% de los mismos muestran angiomatosis leptomeníngea bilateral. Las crisis parciales son generalmente la primera manifestación neurológica y se inician habitualmente en los primeros meses de vida. La TAC craneal puede revelar las calcif!caciones intracraneales características, aunque rara vez están presentes al nacimiento. Presentamos un caso de Sturge Weber con crisis de inicio precoz, nevus facial y calcificaciones intracraneales bilaterales presentes en el periodo neonatal.
Palabras clave: Sturge Weber, angiomatosis leptomeníngea, calcificación intracraneal, nevus facial.
ABSTRACT
The Sturge Weber syndrome is an unfrequent neurocutaneous disorder characterized by the association of a venous angioma of the pia mater with a port–wine stain of the face. Bilateral facial nevi are not uncommon and have been seen in 30% of patients; 25% of these cases have bilateral leptomeningeal angiomatosis. Partial seizures are usually the first neurologic manifestation and frequently begin in the first months of life. CT scan can show the characteristic intracranial calcifications, although they are rarely present at birth. We report a rare case of Sturge Weber disease with an early onset of seizures, bilateral facial nevi and intracranial calcification at the neonatal periodo.
Key words: Sturge Weber, leptomeningeal angiomatosis, intracranial calcification, facial nevi.
El síndrome de Sturge Weber o angiomatosis encéfalo–trigeminal es un trastorno neurocutáneo infrecuente, por lo general esporádico, cuya forma completa se caracteriza por la presencia de angioma pial, angioma venoso facial y angioma coroideol 1,2. Desde el punto de vista clínico, las características del síndrome incluyen las crisis convulsivas, los déficit motores y el retraso mental 3.
La presencia de angiomas faciales bilaterales no supera el 30% de los casos 4. En la cuarta parte de los mismos, se observa un angioma leptomeníngeo también bilateral. Las calcificaciones intracraneales subyacentes al angioma son otra característica neurorradiológica del síndrome. Pueden ser bilaterales en más del 20% de los casos, pero rara vez se observan al nacimiento 5.
Aportamos un caso con síndrome de Sturge Weber que presenta un nevus facial bilateral, crisis convulsivas desde las primeras semanas de vida, y un angioma pial y calcificaciones intracraneales de localización bilateral.
CASO CLÍNICO
Paciente varón de un mes de vida que acude al Departamento de Urgencias por presentar episodios de hipertonía generalizada con tendencia al opistótonos y movimientos clónicos hemifaciales de aproximadamente un minuto de duración. Estos episodios se acompañan de cianosis facial y somnolencia posterior. Aunque están presentes desde hace varios días, se han intensificado en las últimas 48 horas.
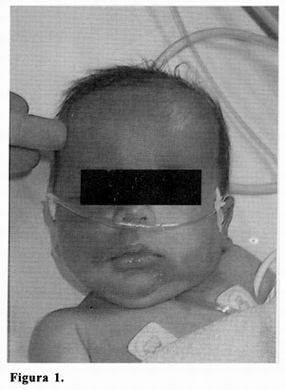
La exploración física revela la presencia de un extenso nevus rojo–vinoso que afecta bilateralmente a la cara, calota, nuca, y alcanza la región pectoral izquierda, respectando de forma parcheada el 1/3 inferior de la hemifacies derecha y el surco nasogeniano izquierdo (figura 1). Se observa igualmente un leve engrosamiento cutáneo fronto–orbitario y nucal. El perímetro craneal es 40 cm (percentil 97). La auscultación pulmonar muestra un patrón respiratorio normal, buena ventilación bilateral y estridor inspiratorio ocasional que se intensifica con el llanto. El examen neurológico manifiesta una leve hipotonía generalizada, con buena respuesta a estímulos, movimientos simétricos y normorreflexia osteotendinosa. La movilidad ocular es adecuada, fija la mirada y sigue objetos aceptablemente.
En los antecedentes personales del niño se refiere un embarazo controlado, gemelar dizigótico. Las ecografías intraútero habían mostrado la presencia de un sistema ventricular supratentorial aumentado de tamaño con marcada alteración de la ecogenicidad cerebral, situación que se confirma eco gráficamente al nacer. Parto a término, eutócico, segundo gemelo; el peso al nacimiento fue de 2870 g; Apgar 6/9; reanimación moderada tipo III. ECO abdominal normal. Los antecedentes familiares no muestran datos relevantes; los padres son jóvenes y sanos; el gemelo no muestra alteraciones clínicas relevantes.
Al ingreso, se realiza estudio EEG que muestra una actividad de fondo deprimida de forma generalizada sobre la que se registran ritmos rápidos de breve duración regiones occipitales derechas y una actividad rítmica infraclínica localizada en región fronto–temporal izquierda con tendencia a la difusión contralateral. Tras el estudio neurofisiológico se inicia tratamiento antiepiléptico con fenobarbital a las dosis habituales; después no se observan nuevas crisis, situación que se mantiene hasta el alta del paciente.
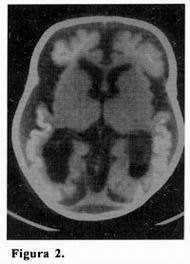
Durante el ingreso se completan los estudios neurorradiológicos. La TAC craneal muestra una moderada atrofia cerebral cortico–subcortical generalizada, predominio izquierdo, con calcificaciones de distribución giral occípito–parietales bilaterales y subcorticales frontales bilaterales (figura 2).
La RM cerebral confirma la atrofía cerebral asimétrica. El contraste evidencia la extensión bilateral del angioma pial (figura 3–a). Se observa igualmente la presencia de vasos agrandados, algunos periventriculares, de naturaleza venosa y patológica (figura 3–b). Las proyecciones sagitales muestran imágenes puntiformes hipointensas a nivel nucal que señalan la alteración cutánea (figura 3–c).



El estudio oftalmológico confirma, en consonancia con el estudio neurorradiológico, la ausencia de alteraciones oculares o angiomas coroideos. El estudio otorrinolaringológico apoya la presencia de una laringomalacia asociada sin alteraciones vasculares añadidas.
DISCUSIÓN
El síndrome de Sturge Weber (SSW), la angiomatosis encefalo–trigeminal o angiomatosis meníngeofacial es un trastorno neurocutáneo infrecuente cuya forma completa se caracteriza por la presencia de alteraciones angiomatosas a nivel leptomeníngeo, ocular y facial 1–4. Afecta a ambos sexos por igual, y aunque la mayor parte de los casos son esporádicos, se han descrito casos heredados de forma autonómica dominante o recesiva 6.
El angioma facial está presente al nacimiento. Afecta generalmente al 1/3 ó 2/3 superiores de la cara, siendo bilateral hasta en el 32% 4,6. La afectación facial bilateral se asocia con una mayor prevalencia de alteraciones intracraneales 7. Contrariamente, el angioma cutáneo puede estar ausente en un 15% de los pacientes con angiomas piales 8,9. Puede extenderse al tronco, observarse también en las extremidades o mucosas.
El angioma ocular se observa en la tercera parte de los casos, afectando a coroides o esclerótica, con una localización ipsilateral al angioma cutáneo. Puede asociarse. de forma evolutiva con glaucoma, cataratas, desprendimiento de retina o alteración de la agudeza visual 6,10.
La patología intracraneal propia del SSW se constituye básicamente por la presencia de un angioma meníngeo limitado por lo general a la piamadre y formado principalmente por vasos venosos dilatados y tortuosos 2–6. Las calcificaciones están presentes en la mayor parte de los pacientes al finalizar la segunda década de la vida 6, afectan en un inicio a la sustancia blanca subcortical y se extiende después hacia la superficie. Se observan rara vez al nacimiento y sólo en el 20% de los casos son bilaterales 5,11–13.
Los estudios neurorradiológicos evidencian estos hallazgos intracraneales. La RNM cerebral con contraste es el método de elección para valorar la extensión del angioma. No es infrecuente observar igualmente una atrofia cortico–subcortical subyacente al angioma, plexos coroideos hiperplásicos e hiperintensos, así como vasos agrandados en regiones subependimarias y periventriculares 14–16. La TAC craneal muestra con mayor precisión la distribución y morfología característica de las calcificaciones, que adquieren forma de serpentina o trayectos irregulares y paralelos 5. Se sitúan generalmente en regiones occipito–parietales, aunque pueden situarse en cualquier localización.
Clínicamente, los problemas asociados más importantes por orden de frecuencia son las crisis epilépticas, el retraso mental y los déficit motores. Las primeras parecen hasta en el 90% de los casos, por lo general los primeros meses de vida 4,6.17 . Pueden ser parciales, generalizadas, mioclónicas, tónicas, atónicas o espasmos infantiles. El control terapéutico es satisfactorio en la mitad de los casos. El trazado EEG intercrítico muestra en las formas unilaterales una actividad de base hipovoltada y asimétrica, con actividad paroxística polimorfa.
El retraso mental se observa en el 40% de los casos 4,6,17. La presencia de crisis o la afectación bilateral empeora la frecuencia y severidad del retraso. La tercera parte de los pacientes con SSW presentan hemiparesia contralateral al angioma, excepto en aquellos pocos casos con afectación vascular bilateral. Aparece generalmente antes de los dos años, tras el inicio de las crisis, aunque rara vez se objetiva en los primeros meses de vida.
El caso referido refleja una condición severa e infrecuente en el SSW. La presencia de un angioma cutáneo bilateral, con la participación angiomatosa intracraneal bilateral, y las calcificaciones extensas subyacentes y presentes al nacimiento, son hallazgos absolutamente infrecuentes en este síndrome. Atendiendo a las frecuencias publicadas aisladamente en relación a estas manifestaciones, tan sólo el 0.6% de los neonatos con SSW van a presentar estas tres alteraciones clínico radiológicas. Es complejo predecir la evolución clínica de este paciente; a priori, la afectación neurológica bilateral, la existencia temprana de una clara atrofia cortico–subcortical, así como la aparición precoz de crisis convulsivas, aunque actualmente controladas, empeora el pronóstico.
REFERENCIAS
1. Sturge WA. A case of partial epilepsy, apparently due to a lesion of one of the vaso–motor centres of the brain. Trans Clin Soc London 1879;12:162–7. [ Links ]
2. Weber FP. Right–sided hemihypertrophy resulting from right–sided congenital spastic hemiplegia, with a morbid condition of the left side of the brain revealed by radiograms. J Neurol Psychopath 1922;3:134–9. [ Links ]
3. Rodríguez–Barrionuevo AC. Síndromes neurocutáneos con predominio de anomalías vasculares. Rev Neurol 1996;24: 1072–84. [ Links ]
4. Pascual–Castroviejo SI, Díaz–González C, García Melian R, González Casado I, Muñoz Hiraldo E. Sturge Weber síndrome: study of 40 patients. Pediatr Neurol 1993; 9:283–8. [ Links ]
5. Gardeur D, Palmieri A, Mashaly R. Cranial computed tomography in the phakomatoses. Neuroradiology 1983;25:293–304. [ Links ]
6. Gómez MR, Bebin EM. Sturge Weber syndrome. En: Gómez MR ed. Neurocutaneous diseases: a practical approach . London: Butterworths 1987. [ Links ]
7. Tallman B, Tan OT, Morelli JG. Location of port–wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics 1991; 87:323–7. [ Links ]
8. Pascual Castroviejo I, Pascual Pascual SI, Viaño J. Sturge Weber síndrome without facial nevus. Neuropediatrics 1995; 26:220–2. [ Links ]
9. Aydin A, Cakmakci H, Kovanlikaya A, Dirik E. Sturge Weber syndrome without facial nevus. Pediatr Neurol 2000; 22: 400–2. [ Links ]
10. Pou–Serradell. Evolución natural de las facomatosis en la edad adulta. Rev Neurol 1996; 24:1085–127. [ Links ]
11. Alonso A, Taboada D, Ceres L, Beltran J, Olague R, Nogues A. Intracranial calcification in a neonate with the Sturge Weber syndrome and additional problems. Pediatr Radiol 1979;8:39–41. [ Links ]
12. Kitahara T, Maki Y. A case of Sturge Weber disease with epilepsy and intracranial calcification at the neonatal periodo Eur Neurol 1978; 17:8–12. [ Links ]
13. Boltshauser E, Wilson J, Hoare RD. Sturge Weber syndrome with bilateral intracranial calcification. J Neurol Neurosurg Psychiatry 1976; 39:429–35. [ Links ]
14. Griffiths PD. Sturge Weber syndrome revisited: the role of neuroradiology. Neuropediatrics 1996; 27:284–94. [ Links ]
15. Benedikt R, Brown D, Walker R, Ghaed V, Mitchell M, Geyer C. Sturge Weber syndrome: cranial MR imaging with Gd–DTPA. AJNR Am J Neuroradiol 1993;14: 409–15. [ Links ]
16. Stimac GK, Sulomon MA, Newton TH. CT and MR of angiomatous malformations of the choroids plexus in patients with Sturge Weber disease. AJNR Am J Neuroradiol 1986; 7: 623–7. [ Links ]
17. Sujansky E, Conradi S. Outcome of Sturge Weber síndrome in 52 adults. Am J Med Genet 1995;57:35–45. [ Links ]

