











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkANTECEDENTES
En la causa de la fiebre de origen desconocido las principales series publicadas a partir de 1990 muestran cambios en la distribución de las distintas causas con disminución de las infecciones y mayor número de casos con enfermedades reumatológicas (inflamatorias no infecciosas). Respecto a las neoplasias, las de origen hematológico están principalmente representadas por el linfoma no Hodgkin.1 La mielofibrosis primaria, por el contrario, generalmente no se manifiesta con fiebre y de hacerlo es de bajo grado. Forma parte de los síndromes mieloproliferativos crónicos y se caracteriza por la proliferación clonal de la célula reticular primaria que secundariamente produce fibrosis de la médula ósea por hiperactividad no clonal de los fibroblastos. En el caso que comunicamos, el padecimiento comenzó como un síndrome febril que reunió los criterios establecidos para definirse como fiebre de origen desconocido clásica, que se acompañaba de importante hepatomegalia y esplenomegalia que hicieron necesaria la realización de laparotomía protocolizada y estudio histopatológico que demostró una metaplasia mieloide megacariopoyética característica.2
CASO CLÍNICO
Paciente masculino de 56 años de edad, que inició su padecimiento seis meses previos a su hospitalización con fiebre diaria de incluso 38.5°C, acompañada de diaforesis nocturna y pérdida de peso de 11 kg, artralgias de manos y pies. Inicialmente fue tratado con vancomicina por sospecha de endocarditis al reportarse en hemocultivo Staphylococcus haemolyticus; sin embargo, la fiebre no remitió. Fue referido a nuestro hospital en donde se descartó vegetación por ecocardiograma transesofágico y se continuó protocolo de estudio por fiebre de origen desconocido.
Se solicitó el resto de cultivos, incluidos para Mycobacterium, coprocultivo y coproparasitoscópico, reacciones febriles, TORCH y panel viral para VIH, VHC y VHB, todos fueron negativos.
A la exploración física se obtuvo lo siguiente: talla: 1.70 cm, peso: 60 kg, IMC: 20.9 kg/m2. Destacó únicamente hepatomegalia no dolorosa 2 cm por debajo del reborde costal, esplenomegalia y ausencia de adenopatías.
Los estudios de laboratorio arrojaron: glucosa 94 mg/dL, Cr 0.90 mg/dL, Na 136.1 mmol/L, Ca 8.1 mg/dL, ALT 129 U/L, AST 116 U/L, hemoglo-bina 8.9 g/dL, hematócrito 29.2%, leucocitos 7.3 K/uL, neutrófilos 55%, linfocitos 20%, monocitos 23%, plaquetas 74,000 K/uL, TP 16.5 seg, INR 1.22, TTPa 32.5 seg, BT 0.57 mg/dL, LDH 260 U/L, GGT 134 U/L, FA 797 U/L, albúmina 2.5g/dL, ácido úrico 5.4. EGO normal.
Marcadores tumorales: ACE 4.05 ng/mL, APE 0.18 (normal), PCR 107 mg/L, VSG 33, IgG 1980 mg/dL (700-1800), IgA 433 mg/dL (100-480), IgM 31.80 (60-250). Hierro 12 μg/dL, captación de hierro 265.4 μg/dL, saturación de hierro 4.52%, transferrina 229 mg/dL. Fibrinógeno 519 mg/dL, lisis de euglobulina > 120 minutos. ANAs y ENAs negativos. Complemento normal.
Estudios de imagen
El ultrasonido Doppler hepático mostró hepatoesplenomegalia. Incremento difuso de la ecogenicidad hepática en sus diferentes segmentos. Derivaciones esplenorrenales. Doppler con cambios parenquimatosos hepáticos difusos e hipertensión portal.
La tomografía computada de abdomen evidenció hepatoesplenomegalia, hígado homogéneo y bazo discretamente heterogéneo. En el retroperitoneo se observaron múltiples imágenes hipodensas en cadenas interaorticocavales, periaórticas en relación con actividad ganglionar (Figura 1).

Figura 1 Tomografía de abdomen que muestra hepatomegalia y esplenomegalia. Actividad ganglionar retroperitoneal.
La panendoscopia mostró várices gástricas IGV1 con datos de mal pronóstico, gastropatía hipertensiva leve de McCormack y xantoma gástrico prepilórico.
Ante el hallazgo de hepatomegalia, esplenomegalia, alteraciones en la citometría hemática (anemia y trombocitopenia) y la existencia de adenomegalias se sospechó un síndrome linfoproliferativo, porque la causa infecciosa se volvió poco probable por los resultados obtenidos y el tiempo de evolución del padecimiento. Por esta razón se tomó la decisión de realizar aspirado de médula ósea y biopsia de hueso. La primera mostró la médula ósea hipercelular con hiperplasia de series mieloide y eritroide y la segunda celularidad global aproximada de 80%, existencia de las tres series hematopoyéticas. Relación mielo/eritroide 8/3 con diferenciación desde promielocitos hasta bandas y segmentados. Reticulocitos de aspecto habitual, algunos con aspecto megaloblastoide. Megacariocitos de 3-4 por campo de alto poder (40X seco fuerte), atípicos, eritrocitos extravasados escasos, fibrosis reticulínica grado I-II focal, médula ósea hipercelular con hiperplasia de serie mieloide y eritroide (Figura 2A).
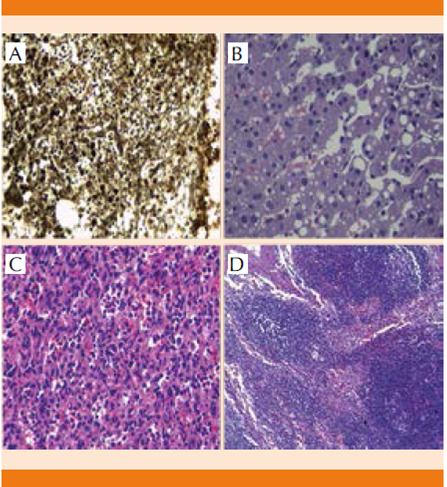
Figura 2 Biopsias. A.Hueso: fibrosis reticulínica grado I-II focal, médula ósea hipercelular con hiperplasia de serie mieloide y eritroide. B. Hígado: Datos histológicos de obstrucción de flujo de salida venoso hepático. C.Bazo: zonas fibrinopurulentas que semejan múltiples abscesos, con hematopoyesis extramedular. D.Ganglio retroperitoneal: hiperplasia mixta sin atipias.
Debido a la falta de conclusión diagnóstica hasta este punto y persistencia de la fiebre, con reportes poco concluyentes y considerando la posibilidad de una enfermedad linfoproliferativa, se programó laparotomía protocolizada que reportó como principales hallazgos ascitis (100 cc), conglomerados ganglionares presacro, intracavoaórticos y periaórticos, hepatomegalia a 4 cm de líneas convencionales, con hígado de apariencia microquística y congestivo y datos histológicos de obstrucción de flujo de salida venoso hepático (Figura 2B). La esplenectomía reportó bazo de 20 x 15 cm con zonas fibrinopurulentas que semejaban múltiples abscesos, adherido a cúpula diafragmática con vasculatura magistral, fibrocongestivo, con periesplenitis y hematopoyesis extramedular (Figura 2C). Ganglios linfáticos con hiperplasia mixta sin atipias (Figura 2D).
La fibrosis reticulínica en la médula ósea, la hematopoyesis extramedular e hiperplasia de series mieloide y eritroide, junto con las alteraciones clínicas en relación con hipertensión portal en hígado no cirrótico y la ausencia de datos histopatológicos compatibles con linfoma apoyaron la sospecha de un trastorno mieloproliferativo como causa de la fiebre de origen desconocido y entre éstos, la mielofibrosis primaria explicaba los datos clínicos, bioquímicos e histopatológicos del paciente al cumplirse dos criterios mayores y tres menores de esa enfermedad.
En la actualidad el paciente recibe tratamiento con talidomida, hidroxiurea y prednisona, con normalización de la citometría hemática, recuperación de peso y remisión de la fiebre.
DISCUSIÓN
En 1961, Petersdorf y Beeson definieron la fiebre de origen desconocido como una temperatura mayor de 38.3°C (101°F) en múltiples ocasiones durante un periodo de tres o más semanas sin obtener el diagnóstico preciso posterior a una semana de estudio con el paciente hospitalizado.3,4 Treinta años después, Durak y Street redefinieron el concepto considerando una fiebre mayor a 38.3°C en múltiples ocasiones, suprimiendo la necesidad de una semana de hospitalización y proponiendo una estancia hospitalaria de sólo tres días o tres consultas médicas subsecuentes, con resultados microbiológicos negativos luego de dos días de incubación. Asimismo, clasificaron la fiebre de origen desconocido en cuatro grupos: clásica, nosocomial, en pacientes neutropénicos y asociada con infección por el virus de la inmunodeficiencia humana (VIH).5 Por último, Knockaert y otros autores propusieron un listado mínimo y más o menos uniforme de estudios diagnósticos negativos para clasificar a un paciente con fiebre de origen desconocido (Cuadro 1).6,7
Cuadro 1 Criterios de fiebre de origen desconocido y estudios mínimos negativos de Knockaert
El diagnóstico de mielofibrosis primaria como causa de fiebre de origen desconocido es excepcional, tiene incidencia de 5-7 casos por millón de habitantes/año. También llamada metaplasia mieloide agnogénica; forma parte de los llamados síndromes mieloproliferativos crónicos. La media de edad de aparición es de 60 a 65 años.8 Evoluciona lentamente y algunos pacientes pueden vivir sin síntomas durante años, 33% de ellos son asintomáticos al momento del diagnóstico.
El signo más frecuente y característico es la esplenomegalia, dato que marcó la pauta en nuestro protocolo diagnóstico y la hematopoyesis extramedular. En 80% de los casos también se observa hepatomegalia. Las adenopatías, raras al inicio de la enfermedad, son algo más frecuentes en los casos de larga evolución en los que también pueden observarse nódulos cutáneos. Las alteraciones bioquímicas más comunes son aumento de LDH, ácido úrico y vitamina B12. Con cierta frecuencia se detectan anticuerpos antinucleares y se observa positividad en la prueba de Coombs.9
En el frotis sanguíneo aparece anemia normocítica normocrómica y los rasgos característicos de la hematopoyesis extramedular: dacriocitos, eritrocitos nucleados, mielocitos y promielocitos; también pueden encontrarse mieloblastos, proceso llamado leucoeritroblastosis. La cifra de leucocitos es muy variable y varía entre leucopenia y leucocitosis intensa. Lo mismo ocurre con las plaquetas porque en 20% de los casos se observa trombocitopenia y en 25% de los casos trombocitosis, esta última alteración fue evidente desde los primeros estudios solicitados a nuestro paciente.
En el aspirado de médula ósea se descubre hipercelularidad medular con hiperplasia de las tres líneas celulares y en particular megacariocitos aumentados, pero sin las típicas alteraciones morfológicas que distinguen a la mielofibrosis idiopática crónica de los otros procesos mieloproliferativos crónicos. La existencia de más de 25% de blastos es consistente con el diagnóstico de mielofibrosis. En las biopsias medulares se observan cuatro lesiones fundamentales: hiperplasia hematopoyética, fibrosis reticulínica, fibrosis colágena y osteoesclerosis.10-12 Los análisis citogenéticos se utilizan para confirmar la mielofibrosis primaria y descartar otros trastornos mieloproliferativos. Entre 50 y 60% de las personas con mielofibrosis primaria tienen una mutación en el gen Janus quinasa 2 (JAK2), causante de la esplenomegalia, anemia y trombocitopenia.13 En nuestro hospital no fue posible realizar este análisis.
La mayoría de los casos (70-80%) se diagnostican en etapa mielofibrótica por la existencia de esplenomegalia acompañada de manifestaciones sistémicas; el restante 20 a 30% se establece en una etapa prefibrótica conocida también como fase celular, como el caso de nuestro paciente. En los casos de mielofibrosis aguda, los pacientes tienen pancitopenia, pero no esplenomegalia. La monocitosis en sangre periférica o médula ósea indican mielodisplasia.14
El objetivo del tratamiento es aliviar los síntomas y reducir el riesgo de complicaciones. La anemia requiere transfusiones, prednisona, eritropoyetina, inmunomoduladores como la talidomida y lenalidomida e incluso terapia con andrógenos (oximetolona y danazol), que puede promover la producción de glóbulos rojos y se prescribe para aliviar los síntomas de la anemia severa. Debe valorarse el requerimiento de esplenectomía en pacientes con hipertensión portal, esplenomegalia dolorosa, infartos esplénicos recurrentes, anemia dependiente de transfusiones o trombocitopenia. Aunque ocasionalmente se observa alivio de la anemia, este procedimiento se asocia con morbilidad de 15 a 30% y con mortalidad de 10% debido sobre todo a infecciones, hemorragias y trombosis. Además, en 20% de los pacientes sobreviene hepatomegalia y trombocitosis después de la esplenectomía.
También se prescribe hidroxiurea o incluso cladribina para tratar la trombocitosis. Los bisfosfonatos pueden aliviar el dolor óseo y mejorar los conteos sanguíneos. El alotrasplante de células madre es la única curación potencial de la mielofibrosis primaria. Sin embargo, por el riesgo notable de efectos secundarios potencialmente mortales no es una opción de tratamiento para la mayoría de las personas con mielofibrosis primaria debido a la edad, el avance de la enfermedad u otros problemas médicos concomitantes. En los últimos años se ha prestado especial interés al desarrollo de inhibidores de la proteína cinasa Jak2. Varios fármacos se encuentran en fase de desarrollo, entre ellos el lestaurtinib.15,16
Los criterios propuestos por la Organización Mundial de la Salud para el diagnóstico de la mielofibrosis exigen los tres criterios principales y dos criterios menores (Cuadro 2).17
Cuadro 2 Criterios diagnósticos de mielofibrosis (OMS)
El pronóstico varía ampliamente entre los pacientes con mielofibrosis primaria. Los factores de riesgo vinculados con el pronóstico de cada paciente se evalúan en forma individual. Mientras que la mediana de la supervivencia para las personas con mielofibrosis primaria es de 3.5 a 5.5 años, las personas menores de 55 años de edad con buenos factores de pronóstico tienen mediana de supervivencia de incluso 11 años. El Sistema Internacional de Puntaje Pronóstico (IPSS) utiliza los siguientes cinco factores de riesgo para estimar la supervivencia a partir del momento del diagnóstico:18
Edad: 65 años o mayor.
Anemia: hemoglobina menor de 10 g/dL.
Síntomas como fiebre, sudoraciones nocturnas o pérdida de peso.
Leucocitosis mayor a 30,000/mL.
Células blásticas sanguíneas circulantes, al menos 1%.
Según el IPSS, los pacientes sin ninguna de estas características adversas, excluida la edad, tienen mediana de supervivencia de más de 10 años. La existencia de por lo menos dos de las características anteriores reduce la mediana de supervivencia a menos de tres años. Alrededor de 12% de los casos de mielofibrosis primaria se transforman en leucemia mieloide aguda, razón por la que este paciente continúa en vigilancia por los servicios de Hematología y de Medicina Interna.19
Con el reporte de este caso queremos destacar que la mielofibrosis primaria es una causa excepcional de fiebre de origen desconocido y se diagnostica con base en una serie de criterios bien establecidos y alteraciones principalmente del sistema hematopoyético. Para establecer el diagnóstico etiológico temprano y otorgar el tratamiento oportuno debe seguirse un protocolo de estudio ordenado y sistematizado según los recursos de cada hospital para descartar los diagnósticos diferenciales de este síndrome, destaca el papel de la laparotomía exploradora y la toma de biopsias.

