











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkANTECEDENTES
El glomérulo renal, estructura altamente vascularizada, se ve afectado por vasculitis de pequeño vaso asociada con anticuerpos anticitoplasma de neutrófilo (ANCA), descrita por su relativa escasez de depósitos inmunes como pauciinmune. Estas vasculitis se clasifican según del consenso internacional de Chapel-Hill en poliangeítis microscópica, granulomatosis con poliangeítis, granulomatosis eosinofílica y la llamada vasculitis limitada a riñón.1
Los dos principales autoanticuerpos implicados en la patogénesis de la glomerulonefritis paucinmune son los dirigidos a antígenos citoplasmáticos de neutrófilos y monocitos: proteinasa-3 (PR3) y mieloperoxidasa, descritos por su participación como citoplasmático, C-ANCA, y perninucleares, P-ANCA.
La vasculitis asociada con ANCA genera afección multisistémica y en un gran porcentaje de los pacientes lesión renal grave de rápida evolución, proteinuria y hematuria glomerular, síndrome nefrológico llamado glomerulonefritis rápidamente progresiva. La biopsia renal es el patrón de referencia diagnóstico, además de proporcionar valor pronóstico. Desde el punto de vista anatomopatológico la glomerulonefritis rápidamente progresiva puede dividirse en pauciinmune, antimembrana basal glomerular (GBM GN) y mediada por inmunocomplejos. El tratamiento en general es con inmunosupresor y terapia renal de soporte desde medidas conservadoras hasta tratamiento de sustitución renal. Se comunica un caso de glomerulonefritis rápidamente progresiva asociada con ANCA por confirmación anatomopatológica.2
CASO CLÍNICO
Paciente masculino de 70 años de edad. Antecedentes de importancia: nefrectomía derecha (1972) secundaria a quiste renal simple (reporte histopatológico benigno), arritmia no especificada (2014) en tratamiento con propafenona y rivaroxaban e hiperplasia prostática benigna (2014), por lo que le realizaron resección transuretral de próstata, sin tratamiento. Inició su padecimiento hacía dos meses con odinofagia, rinorrea hialina, congestión nasal y cefalea frontal; se estableció el diagnóstico de rinosinusitis. Se agregó edema bilateral de ambos miembros inferiores. Se reportó hiperazoemia con criterios de diálisis. Recibió 11 sesiones de hemodiálisis, sin abordaje alguno. Acudió a valoración hospitalaria. Se hicieron estudios de laboratorio generales en los que se observó elevación de azoados (Cr 3.76, BUN 38), examen general de orina con proteinuria, albuminuria, leucocituria y hematuria, con radio albúmina/creatinina 1653 mg/g. Se inició abordaje de glomerunefritis de origen a determinar; se realizaron estudios complementarios en los que destacó complemento C3 normal, con C4 bajo ANAs positivo, anticuerpos mieloperoxidasa positivos, ante la sospecha de vasculitis sistémica asociada con ANCA se realizó biopsia renal, con resultado de glomerulonefritis pauciinmune, con lo que se estableció el diagnóstico de vasculitis asociada con ANCA. Se inició tratamiento con metilprednisolona durante tres días (1 g intravenoso) y rituximab. Los Cuadros 1 y2 muestran los estudios de laboratorio completos.
Cuadro 1 Resultados de las pruebas de laboratorio
| HB | 9.7 g/dL |
| ALB | 2.7 g/dL |
| Cr | 3.76 mg/dL |
| BUN | 38 mg/dL |
| PCR | 179.2 |
| ANAs | 1: 80 |
| C4 | 15.5 mg/dL |
| C3 | 92 mg/dL |
| MPO | 239.8 AI |
| PR3 | (-) |
Cuadro 2 Resultados del examen general de orina
| Apariencia | Turbia |
| Cilindros | (-) |
| Hemoglobina | 1+ |
| Leucocitos | 835 Xc |
| Eritrocitos | 77 Xc |
| Proteinuria | 201 mg/dL |
| Creatinuria | 44.4 mg/dL |
| Albuminuria | 73.4 mg |
| Radio alb/Cr | 1,653 mg |
| Radio prot/Cr | 4.5 g |
En la recolección de orina de 24 horas se observó proteinuria en rango subnefrótico: proteínas totales orina de 24 horas: 1611 mg/24 horas, electroforesis de proteína de 24 horas, en el que no se observaron picos monoclonales, con resultado de proteínas totales de: 1469 mg/24 horas, albúmina: 51.5%, alfa 1: 7.8%, alfa 2: 10.9%, beta: 16.3%, gamma: 13.5%.
El paciente alcanzó cifra máxima de creatinina de 6.18 mg/dL y durante el segundo día de administración de esteroide intravenoso la cifra de creatinina alcanzó valores de 3.1 mg/dL, sin más requerimientos dialíticos.
DISCUSIÓN
Epidemiología
Las vasculitis asociadas con ANCA afectan a cualquier edad, sobre todo en la edad adulta y ambos sexos de manera similar. La más frecuente en nuestro país es la poliangeítis microscópica con incidencia anual de 8 casos por millón de habitantes, seguida de la granulomatosis con poliangeítis con 3 casos por millón de habitantes y el síndrome de Churg-Strauss con 1.3 casos por millón.3
La incidencia es mayor en el sexo masculino con incidencia anual global de 10-20 millones con pico entre 65 y 74 años de edad en todo el mundo. La prevalencia es de alrededor de 32 casos por millón solamente en Estados Unidos en comparación con Europa donde la prevalencia corresponde a 65 casos por millón. La incidencia es mayor en caucásicos que en los afroamericanos con riesgo dos veces mayor de vasculitis asociada con ANCA en los pacientes con ascendencia europea.4
El 80% de los casos de glomerulonefritis rápidamente progresiva de tipo pauciinmune se relaciona con granulomatosis con poliangeítis ANCA-positivo, 90% se relacionan con poliangeítis microscópica ANCA-positiva y sólo 50% se relacionan con granulomatosis eosinofílica.5,6
Fisiopatogenia
La glomerulonefritis rápidamente progresiva es un síndrome caracterizado por pérdida rápida de la función renal, en semanas o meses, incluye eritrocituria dismórfica y proteinuria glomerular, síndrome catalogado como síndrome nefrítico. Se asocia con existencia de semillas en 50% o más de los glomérulos, de manera que el término glomerulonefritis rápidamente progresiva es equivalente al término glomerulonefritis semilunar. Investigaciones recientes identificaron otros anticuerpos asociados con la glomerulonefritis rápidamente progresiva, por lo que se entiende como un espectro complejo de la enfermedad, con considerable sobrelapamiento en términos de genotipo clínico y desenlaces de la enfermedad. Además, muchos factores genéticos y ambientales se han implicado recientemente en la patogénesis de esta enfermedad.
Esta enfermedad es mediada por mecanismos inmunológicos, a pesar de la escasez de depósitos inmunes, con múltiples modelos animales de enfermedad renal asociada con ANCA, entre los que destacan el de Xiao y su grupo, en el que se inyectaron esplenocitos de un ratón inmunizado contra mieloperoxidasa (MPO) a ratones depletados de células B y T y a ratones tipo salvaje, resultando en la aparición de glomerulonefritis pauciinmune necrotizante semilunar y capilaritis pulmonar hemorrágica. En términos histopatológicos es idéntico al genotipo de la vasculitis humana asociada con MPO-ANCA.7 Los ANCA son autoanticuerpos dirigidos contra proteínas específicas en el citoplasma de los neutrófilos y monocitos, la mieloperoxidasa (MPO) o la proteinasa 3 (PR3) son los principales antígenos en pacientes con vasculitis y glomerulonefritis. Después de la unión con los antígenos el resultado es la activación de los neutrófilos y monocitos; una vez que la célula ha sido expuesta a dosis bajas de factor de necrosis tumoral alfa, interleucina 1 e interleucina 18, que resulta en la expresión de superficie de PR3 y mieloperoxidasa, permitiendo la interacción con ANCA. La activación del neutrófilo está mediada por la unión de los receptores Fcg y Fab 2.8,9 La activación de los neutrófilos mediada por ANCA resulta en la producción de especies reactivas de oxígeno y en la liberación de enzimas líticas como la elastasa, que son nocivas para las células endoteliales,10-12 así como en la liberación de trampas extracelulares de neutrófilos, compuestas de fibras de cromatina y autoantígenos, incluyendo mieloperoxidasa y PR3, que pueden dañar las células endoteliales y el glomérulo, activar el complemento y contribuir con la propagación de la respuesta inmunitaria por ANCA.13,14 El daño asociado con PR3-ANCA se vincula con inflamación granulomatosa, mayor daño extrarrenal y mayor tasa de recaída, mientras que el daño asociado con MPO-ANCA es más frecuentemente limitado al riñón, con peor pronóstico renal. Se ha observado también la relación con factores ambientales en la patogénesis de la glomerulonefritis rápidamente progresiva, donde los factores microbianos juegan un papel importante en el inicio de la respuesta autoinmunitaria, por el mimetismo con antígenos del hospedero, lo que se ha observado, por ejemplo, en portadores crónicos de Staphylococcus aureus y en bacterias gramnegativas en el contexto de urosepsis.13,14
Hay asociación con algunos HLA; HLA-DP1 y granulomatosis con poliangeítis, HLA-DQ y poliangeítis microscópica y HLA-DRB4 y granulomatosis eosinofílica con poliangeítis.
Además de la producción de autoanticuerpos, las células B tienen la capacidad de presentar antígenos a las células T y pueden modular la respuesta de las células T en las vasculitis de pequeño vaso; se ha demostrado que hay reducción en el número de Bregs en la vasculitis asociada con ANCA.15
Cuadro clínico
En el curso clínico de esta enfermedad destacan dos fases importantes de acuerdo con el periodo de manifestación:
Fase temprana (pre-vasculitis): se caracteriza por síntomas de atopia, dermatitis en los primeros años del curso de la enfermedad, posteriormente alrededor de la edad de 30-35 años se puede agregar asma y rinitis alérgica de difícil control, el rasgo más característico de esta etapa es la infiltración eosinofílica en los tejidos.
Fase intermedia o fase vasculítica: se caracteriza por daño a pequeñas arterias, vénulas y capilares, además de granulomas vasculares y extravasculares, las manifestaciones clínicas de esta fase son inespecíficas; sin embargo, se encuentran en 78-90% de los pacientes (fiebre, pérdida de peso, astenia y adinamia). El resto de las manifestaciones clínicas dependen de la extensión y sitios de lesión vascular y extravascular, es decir, el órgano afectado.16
Pulmonar: entre 50 y 90% de los pacientes tienen manifestaciones pulmonares caracterizadas por asma de difícil control; en 50-60% de los pacientes hay infiltrados eosinofilicos en el pulmón y en reportes de casos incluso 8% de los pacientes tuvieron tromboembolismo pulmonar.17
Cardiovasculares: puede haber pericarditis en alrededor de 25 a 36% de los pacientes. Sin embargo, hasta en 67% de los pacientes pueden encontrarse cambios en el electrocardiograma (onda T), así como miocarditis o insuficiencia cardiaca en 32 a 50%.18
Neurológicas: la manifestación más frecuente que se reporta en 75% de los pacientes es la neuropatía periférica (mononeuritis múltiple) que puede llegar a ser incapacitante.
Renal: el espectro clínico de la enfermedad se resume en la aparición de glomerulonefritis rápidamente progresiva, que se caracteriza por disminución de la tasa de filtración glomerular de, incluso, 50% en un periodo de semanas hasta tres meses, además de hematuria microscópica o macroscópica y proteinuria en rango no nefrótico. Alrededor de 60% de los pacientes necesitan terapia de sustitución renal al momento del diagnóstico.19
Abordaje diagnóstico
Debido a que es una enfermedad multisistémica, la vasculitis en un órgano específico debe ser la sospecha clínica pivote en un paciente con lesión renal grave (por ejemplo, púrpura como afección cutánea o mononeuritis múltiple como afección al sistema nervioso central). El daño renal es de las manifestaciones de más trascendencia en cuanto a la evolución y pronóstico, en poliangeítis microscópica se manifiesta en 90%, 80% en granulomatosis con poliangeítis y 45% en granulomatosis eosinofílica.20 La afección pulmonar, además de en otros órganos y sistemas en específico, se muestra en el Cuadro 3.
Cuadro 3 Frecuencia de la afección al riñón y a otros órganos
| Órgano afectado | Poliangeítis microscópica (%) |
Granulomatosis con poliangeítis (%) |
Granulomatosis eosinofílica (%) |
|---|---|---|---|
| Pulmón | 50 | 90 | 70 |
| Sistema gastrointestinal | 50 | 50 | 50 |
| Piel | Variable | Variable | Variable |
| Sistema nervioso central | 30 | 50 | 70 |
| Vías respiratorias altas | 35 | 90 | 50 |
El paciente suele padecer síntomas constitutivos, como malestar general, artralgias o fatiga, la lesión renal aguda puede ser poco específica, aunque puede estar presente como volúmenes urinarios disminuidos, sobrecarga hídrica o alteraciones sutiles en la orina, orina espumosa, hematuria. En cuanto a la evaluación de la lesión renal, la proteinuria mayor de 1 g/día, hematuria glomerular (eritrocitos dismórficos o cilindros eritrocitarios)21 y FE NA < 1% sin existencia evidente de depleción de volumen pueden orientar el diagnóstico a una causa glomerular. El siguiente paso, una vez establecida la presencia o sospecha de vasculitis y lesión renal aguda, será la medición de ANCAs séricos, realizada a través de inmunofluorescencia indirecta o ELISA. Los títulos de c-ANCA más comúnmente se relacionan con granulomatosis con poliangeítis (75%) y en menor proporción con granulomatosis eosinofílica (5%), mientras que los títulos de p-ANCA se asocian más con vasculitis limitada al riñón (70%) y menos con granulomatosis con poliangeítis (20%). Reconocer la ausencia de éstos incluso en 10-20% de los pacientes es importante para no desviar el diagnóstico y la existencia de ambos, aunque es sumamente rara, suele aparecer en caso de infección o vasculitis asociada con fármacos. Hace poco se vinculó la aparición de anticuerpos asociados con lisosoma de proteína transmembrana 2 (LAMP-2) en glomerulonefritis asociada con ANCA como tercer anticuerpo relacionado con la patogénesis de esta enfermedad.
Otro dato importante a considerar es la manifestación del síndrome nefrítico; en esta enfermedad renal es rápidamente progresivo. La evaluación del complemento juega también un papel importante en el abordaje diagnóstico, generalmente éste está bajo en glomerulonefritis mediada por inmunocomplejos.2 Una vez realizada la biopsia, deberá realizarse la interpretación con un nefropatólogo experimentado porque, además del diagnóstico definitivo, aportará información acerca del pronóstico. Las características generales son necrosis capilar, proliferación extracapilar, infiltrados periglomerulares e intersticiales, arteritis necrotizante y la mencionada ausencia o escasez de inmunocomplejos. Se han descrito cuatro variedades anatomopatológicas asociadas de glomerulonefritis por ANCA: focal, crescéntica, mixta y esclerótica.22
Tratamiento
El tratamiento de la vasculitis asociada con ANCA puede dividirse en tres fases: inmunosupresión, mantenimiento y recaída.3
El tratamiento de la granulomatosis con poliangeítis y la poliangeítis microscópica debe prescribirse en función de la extensión y gravedad de la enfermedad. Existen cinco categorías definidas por el Grupo de Estudio Europeo de Vasculitis (EUVAS): 1. Enfermedad localizada. 2. Enfermedad sistémica temprana (excepto renal). 3. Enfermedad generalizada (amenaza orgánica) Cr. < 5.7 mg/dL. 4. Enfermedad con afectación renal grave (creatinina > 5.7 mg/dL). 5. Enfermedad resistente.
Las categorías 3 y 4 relacionadas con daño renal y, por ende, con glomerulonefritis asociadas con ANCA se tratan de manera distinta. En la categoría 3 (enfermedad generalizada) se prefiere la prednisona a dosis de 1 mg/kg/día junto con ciclofosfamida en pulsos endovenosos (15 mg/kg) cada dos y tres semanas y posteriormente mensuales hasta conseguir la remisión (mínimo de seis pulsos), continuar con prednisona a dosis de 1 mg/kg/día durante cuatro semanas y disminuir progresivamente. En la categoría 4 (afectación renal grave) se incluyen bolos de metilprednisolona en pulsos de 1 g diario (250 mg/6 h) durante tres días y continuar con la dosis habitual de prednisona de 1 mg/kg/día, ciclofosfamida en pulsos endovenosos igual que en la categoría 3 y recambios plasmáticos (mínimo siete recambios plasmáticos) además de la administración de inmunoglobulinas endovenosas a dosis de 200 mg/kg, cada dos recambios plasmáticos. En caso de deterioro renal progresivo, a pesar del tratamiento sistémico administrado, se procederá a iniciar sesiones de hemodiálisis.
Las asociaciones EULAR-EDTA formularon nuevas recomendaciones para el tratamiento de las vasculitis asociadas con ANCA en 2016, algunas en relación con daño renal, entre las que destacan:
Para la inducción de la remisión de vasculitis asociada con ANCA (AVV) de nueva aparición que ponga en peligro órganos vitales o amenace la vida se recomienda el tratamiento con la combinación de glucocorticoides y ciclofosfamida o rituximab.23
Para la inducción de la remisión de vasculitis asociada con ANCA que no amenace la vida o la función de los órganos vitales se recomienda el tratamiento con la combinación de glucocorticoides y metotrexato o micofenolato de mofetilo.23
El intercambio de plasma debe considerarse en los pacientes con vasculitis asociada con ANCA y concentración de creatinina sérica ≥ 500 mmol/L (5.7 mg/dL), debido a glomerulonefritis rápidamente progresiva en el contexto de una enfermedad nueva o recidivante.23
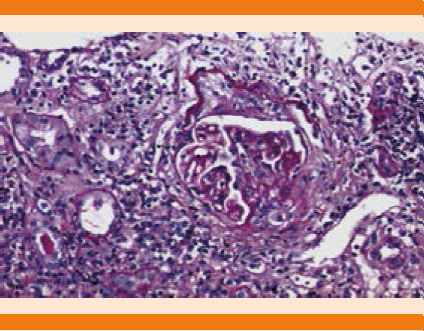
Figura 1 Descripción microscópica: túbulos con edema del epitelio, cilindros hialinos y hemáticos, atrofia y fibrosis intersticial.
El recambio plasmático puede ser benéfico en tres condiciones:31) hemorragia pulmonar, 2)insuficiencia renal dependiente de diálisis; 3)enfermedad anticuerpos antiglomérulo de membrana basal concurrente.
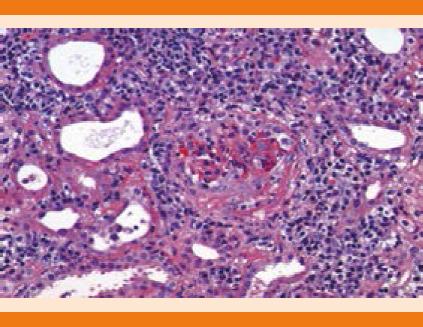
Figura 2 Descripción microscópica: proliferación extracapilar con formación de medias lunas fibrocelulares, infiltrado linfoplasmocitario, neutróficos y eosinófilos.
Con estas nuevas recomendaciones se destaca la administración de rituximab como tratamiento innovador y en algunos lugares como de primera elección debido a sus excelentes resultados en comparación con los tratamientos pasados.
Pronóstico
La vasculitis asociada con ANCA se relaciona con mortalidad y morbilidad altas. La enfermedad renal en etapa terminal se manifiesta a cinco años incluso en 20% de los pacientes con vasculitis asociada con ANCA. Estudios prospectivos han demostrado que con las nuevas terapias farmacológicas la remisión de la enfermedad es alcanzable, incluso, en casi 90% de los pacientes por seis meses y la tasa de supervivencia es de alrededor de 75%. En la actualidad la tasa de recaída continúa siendo alta, hasta de 50% en cinco años a pesar del tratamiento; el mismo tratamiento confiere en la mayoría de los pacientes alto riesgo de complicaciones infecciosas.2
Asimismo, Lionaki y colaboradores demostraron que a pesar de los tratamientos existentes contra la vasculitis asociada con ANCA, una cuarta parte de los pacientes padecerán enfermedad renal terminal.6
Un estudio retrospectivo publicado en 2013 analizó los factores de pronóstico en 273 pacientes consecutivos que fueron diagnosticados con vasculitis asociada con ANCA. En los pacientes con lesión renal grave la necesidad de reemplazo renal se relaciona con peor pronóstico. El análisis multivariado reveló que los principales determinantes de la supervivencia a largo plazo fueron la función renal a seis meses y recaídas renales.3
CONCLUSIONES
El reconocimiento del daño renal de las vasculitis asociadas con ANCA cobra importancia particular debido a su rápida progresión y valor pronóstico. El abordaje de este tipo de enfermedades conlleva al clínico a considerar causas múltiples de deterioro renal; sin embargo, el interrogatorio y la exploración física detallados y las pruebas de funcionamiento renal deben ser los pilares en cuanto el reconocimiento y el rápido tratamiento en estos pacientes con el fin de preservar la función renal y la calidad de vida.

