











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkANTECEDENTES
El síndrome hemolítico urémico es una microangiopatía trombótica caracterizada por anemia hemolítica microangiopática, trombocitopenia y daño renal agudo.1,2 El síndrome hemolítico urémico típico (el más común) es ocasionado por bacterias productoras de la toxina Shiga, típicamente por cepas de Escherichia coli, éste suele aparecer luego de un episodio de diarrea sanguinolenta por estos agentes y en pocos pacientes evoluciona a daño renal agudo.2,3 El término síndrome hemolítico urémico atípico se usa para referirse a los pacientes que padecen este cuadro por causas diferentes, es la forma menos frecuente, representa únicamente 10% de los casos.1-4
El síndrome hemolítico urémico atípico es causado casi siempre por uno o varios defectos en la regulación de la vía alterna del complemento,5 secundario en 60% de los pacientes a anormalidades genéticas o adquiridas en factores reguladores o activadores de esta vía;5,6 sin embargo, algunos pacientes también pueden tener mutaciones en moléculas que no están directamente relacionadas con el sistema del complemento, entre las que destacan la enzima diacilglicerol quinasa,7 el plasminógeno y el factor XII de la coagulación.8
El síndrome hemolítico urémico atípico, a pesar de ser poco frecuente, se asocia con complicaciones y secuelas importantes, secundarias a alteraciones en la función renal; puede cursar con trombocitopenia severa (< 50,000/mL plaquetas) en 42% de los niños y en 27% de los adultos.9 De igual forma, el daño renal agudo asociado es severo en 59% de los niños y 81% de los adultos y puede acompañarse de afectación glomerular con elevación de las concentraciones de creatinina, hematuria, proteinuria, edema e hipertensión.10 Además, en 20 a 48% de los pacientes pueden ocurrir manifestaciones extrarrenales, como alteración del estado mental, convulsiones, pancreatitis, diarrea y hepatitis.10,11
Desafortunadamente hoy día no existe una prueba definitiva para establecer el diagnóstico preciso de síndrome hemolítico urémico atípico, por lo que éste se considera un diagnóstico de exclusión,1 porque ante la sospecha clínica de microangiopatía trombótica deben descartarse primero otras causas más frecuentes, como síndrome hemolítico urémico típico y la púrpura trombocitopénica trombótica.12 Para identificar la mutación genética subyacente es necesario realizar pruebas genéticas costosas; sin embargo, esta caracterización genética más que importancia diagnóstica, tiene gran valor pronóstico y permite prescribir el tratamiento adecuado y disminuir significativamente la morbilidad y mortalidad atribuibles a esta condición. Esta revisión tiene como propósito tratar aspectos generales del enfoque del paciente con síndrome hemolítico urémico atípico, con insistencia en las bases genéticas de esta enfermedad y la utilidad del perfil genético en estos casos.
Epidemiología
El síndrome hemolítico urémico es una afección rara con incidencia global de 1 a 2 casos por cada 100,000 habitantes y sólo 10% se clasifican como síndrome hemolítico urémico atípico.13
El primer episodio de síndrome hemolítico urémico atípico suele manifestarse en población adulta; sin embargo, también puede afectar a niños usualmente antes de los dos años de edad.9 Una condición frecuentemente relacionada con esta enfermedad es el embarazo, se encuentra en 20% de todos los casos de síndrome hemolítico urémico atípico;14 es uno de los principales desencadenantes y representa el mayor número de casos en mujeres;9 esta complicación puede identificarse en 1 de cada 25,000 embarazos15 y la mayoría sobreviene durante el puerperio (75%).16
Fisiopatología
La lesión responsable del cuadro característico del síndrome hemolítico urémico atípico es la microangiopatía trombótica, caracterizada por trombos plaquetarios en la microcirculación;17 los microtrombos crean superficies dentadas en los vasos que ocasionan daño de los glóbulos rojos a medida que éstos los atraviesan, éste es el mecanismo responsable de la anemia hemolítica (anemia hemolítica microangiopática) y de los cambios característicos en la morfología eritrocitaria relacionados con esta condición (esquistocitos).12
A través del tiempo se ha establecido una asociación entre el síndrome hemolítico urémico atípico y la activación incontrolada del complemento debido a mutaciones genéticas; aproximadamente 50% de los pacientes tienen mutaciones heterocigóticas de pérdida de función en genes que codifican inhibidores de la vía alterna, como: factor H (CFH), factor I, proteína cofactor de membranas (MCP), proteínas relacionadas con CFH (CFHR), proteína de unión y C4b (C4bBP);18-20 existen otras mutaciones de ganancia de función en los genes que codifican el factor B (CFB) y C3,18-20 que promueven la activación de esta vía. Además, 3 a 12% de los pacientes tienen mutaciones en más de un gen del complemento.21 Las mutaciones más frecuentes son las que afectan el gen del complemento o autoanticuerpos del factor H, incluso 70% de los pacientes se identifican con síndrome hemolítico urémico atípico.22
Asimismo, a pesar de ser una entidad de origen genético, existen factores ambientales vinculados con su aparición21 porque estas mutaciones requieren un evento desencadenante para producir la enfermedad.5 Los eventos infecciosos desencadenan el síndrome en 50 a 80% de los pacientes, especialmente las infecciones de las vías respiratorias superiores, de igual forma, los cuadros de diarrea aguda bacteriana pueden preceder la aparición del síndrome hemolítico urémico atípico.23 Asimismo, en mujeres, el embarazo, particularmente el periodo posparto, es un factor desencadenante de síndrome hemolítico urémico atípico;14-46 también puede aparecer el síndrome debido a trastornos o defectos en el metabolismo de la cobalamina, debidas principalmente a infecciones por el virus de la inmunodeficiencia humana (VIH), malignidades, consumo de inhibidores de calcineurina, trasplante y enfermedades reumatológicas.24 Por tanto, se considera que otros factores parecen ser necesarios para superar el umbral de estrés tolerado por el endotelio que conduce a lesiones severas de microangiopatía trombótica.5
A partir de los nuevos descubrimientos, como las mutaciones en el gen DGKE que codifica la diacilglicerol cinasa-ε, se ha considerado una definición más amplia para el diagnóstico de síndrome hemolítico urémico atípico, porque esta molécula parece no tener un papel claro en la regulación del complemento; más bien, forma parte de la vía de coagulación y su deficiencia se manifiesta como un estado protrombótico que lleva a microangiopatía trombótica.7
Manifestaciones clínicas
La mayoría de los pacientes con síndrome hemolítico urémico atípico muestran la tríada clásica de anemia hemolítica microangiopática, trombocitopenia y daño renal agudo;4 el cuadro es de inicio abrupto en la mayoría de los casos; sin embargo, en 20% de los pacientes éste puede tener un curso insidioso,25 con anemia subclínica, trombocitopenia fluctuante y función renal conservada.26
En las formas agudas, la anemia hemolítica microangiopática puede manifestarse con palidez, fatiga, soplo sistólico y taquicardia.12 Asimismo, aunque la trombocitopenia puede ser marcada, el sangrado espontáneo y las petequias son poco frecuentes;12 el daño renal agudo puede darse por afectación glomerular y se manifiesta con edema, proteinuria, hematuria y síntomas de hipertensión, como cefalea, convulsiones y otros síntomas urémicos, como astenia, anorexia y emesis.12
El 20 a 48% de los pacientes pueden cursar con manifestaciones extrarrenales,11 que incluyen: manifestaciones cardiacas (3-10%) agudas y crónicas;9 la hipertensión arterial, el infarto agudo de miocardio, la insuficiencia cardiaca y las lesiones vasculares estenosantes son las más frecuentes,11 las anomalías neurológicas pueden ocurrir por afectación microangiopática o por la uremia y son más frecuentes en niños, se manifiestan en 16% de los casos, cursando con alteración en el estado mental, convulsiones, déficits focales (disartria, afasia y disfagia),9 agitación psicomotora, infarto cerebral y coma. Asimismo, cuando hay afectación del sistema gastrointestinal puede sobrevenir dolor abdominal y vómito y, en algunos casos, pancreatitis, diabetes mellitus, isquemia intestinal y hepatitis6,11,27 con elevación de las transaminasas.27
Pueden ocurrir otras alteraciones poco frecuentes y con pocos casos reportados, como afectación ocular,28 que cursa con edema de disco óptico y oftalmoplejía, y afectación de la piel;29 a partir de este hallazgo en la actualidad se está estudiando la importancia de la biopsia de piel para el diagnóstico de la enfermedad.29
Hallazgos de laboratorio
La hemólisis intravascular puede manifestarse con elevación de la deshidrogenasa láctica (LDH), valores de hemoglobina < 10 g/dL, concentraciones bajas de haptoglobina y esquistocitos y reticulocitos en el frotis de sangre periférica,2,12,25 además, la prueba de Coombs suele ser negativa, lo que descarta un origen autoinmunitario de la hemólisis.25)
La trombocitopenia severa con recuento plaquetario por debajo de 50,000 es común que se manifieste en 42% de los niños y en 27% de los adultos, pero 15% de los pacientes pueden tener recuento plaquetario normal.9
Asimismo, las concentraciones séricas de C3 son bajas en 35.9% de los pacientes, esto puede ocurrir debido a la existencia de una mutación directa en el gen del C3 o en mutaciones en los genes que codifican los factores B y H del complemento.9-30
En cuanto a la función renal, puede manifestarse como un síndrome nefrítico, la hematuria y la proteinuria son los hallazgos más comunes,30,31 además de una marcada elevación del nitrógeno ureico (BUN) y la creatinina;10,12 de igual forma, algunos pacientes pueden padecer síndrome nefrótico.31,32 Del mismo modo, los pacientes muestran alteraciones hidroelectrolíticas, como hipercalemia, acidosis metabólica e hiperfosfatemia debido al daño renal.12
Enfoque diagnóstico
Cuando existe la sospecha clínica de estar ante un caso de microangiopatía trombótica deben considerarse tres diagnósticos diferenciales principales: síndrome hemolítico urémico típico, púrpura trombocitopénica trombótica y síndrome hemolítico urémico atípico.12 En los pacientes con manifestaciones clínicas que encajan entre estos posibles diagnósticos es necesario realizar una historia clínica completa que permita identificar factores ambientales desencadenantes y que incluya antecedentes personales y familiares porque un antecedente familiar de microangiopatía trombótica sugiere en gran medida la existencia de síndrome hemolítico urémico atípico;9 además de un examen físico detallado que revele signos y síntomas que el paciente aún no haya manifestado.
Anteriormente el predominio de la afección renal en el síndrome hemolítico urémico y el daño de la función neurológica en la púrpura trombocitopénica trombótica distinguían estas dos enfermedades. Sin embargo, en la actualidad se ha descrito que 50% de los pacientes con púrpura trombocitopénica trombótica tienen alteración de la función renal y 50% de los pacientes con síndrome hemolítico urémico atípico tienen alteraciones neurológicas, lo que evidencia que las características clínicas, a pesar de ser de gran utilidad en el diagnóstico diferencial, pueden solaparse en estas dos enfermedades.26,33 De igual manera, las manifestaciones clínicas tampoco permiten diferenciar entre síndrome hemolítico urémico típico y atípico, porque hasta 30% de los pacientes con síndrome hemolítico urémico atípico iniciarán con el síndrome tras un cuadro diarreico que típicamente es característico de síndrome hemolítico urémico típico.22,23,26
Desafortunadamente en la actualidad no existe una prueba definitiva para establecer el diagnóstico de síndrome hemolítico urémico atípico, por lo que éste se considera un diagnóstico de exclusión,1 pero algunos estudios paraclínicos, como el recuento plaquetario y el grado de afectación renal pueden ser de gran ayuda al momento de orientar el diagnóstico hacia alguna de estas enfermedades;26 por ejemplo, la púrpura trombocitopénica trombótica adquirida cursa en 73% con trombocitopenia grave < 20,000/mm3 y moderada afectación renal, mientras que el síndrome hemolítico urémico atípico suele cursar con trombocitopenia moderada (50-100,000/mm3) y grave afectación renal.26-34 Lo anterior es útil para orientar el diagnóstico en las primeras horas de ingreso del paciente, pero la determinación de la actividad de ADAMTS13 y la prueba de la toxina Shiga resultan esenciales para establecer el diagnóstico preciso entre púrpura trombocitopénica trombótica, síndrome hemolítico urémico típico y síndrome hemolítico urémico atípico.26 En el Cuadro 1 se listan las pruebas diagnósticas específicas para cada una; la púrpura trombocitopénica trombótica es causada por una deficiencia congénita o adquirida de la proteína ADAMTS13, encargada de escindir al factor de Von Willebrand (FVW);35 las formas congénitas de púrpura trombocitopénica trombótica son raras y son causadas por mutaciones homocigotas o dobles heterocigotas del gen ADAMTS13, mientas que las formas adquiridas sobrevienen por la producción de autoanticuerpos (IgG, IgA o IgM) contra esta enzima que ocasionan que su actividad en plasma disminuya; por tanto, una actividad inferior a 5-10% de esta proteína indica púrpura trombocitopénica trombótica adquirida, mientras que valores superiores a 5-10% de actividad de ADAMTS13 aparecen fundamentalmente en el síndrome hemolítico urémico atípico.35,36 Phillips y su grupo recomiendan que una actividad de ADAMTS13 > 10% en un paciente con microangiopatía trombótica debería requerir un examen genético para detectar anormalidades en el complemento37 y un posible síndrome hemolítico urémico atípico y mientras se realizan los estudios genéticos el paciente debe recibir tratamiento contra síndrome hemolítico urémico atípico;38 de la misma manera, la detección de la denominada toxina Shiga por técnicas de cultivo microbiológico, PCR, pruebas serológicas para la detección de anticuerpos contra estas toxinas o una prueba comercial rápida positiva en pacientes con microangiopatía trombótica es diagnóstica de síndrome hemolítico urémico típico.35 En la Figura 1 se muestra el protocolo diagnóstico del síndrome hemolítico urémico.39
Cuadro 1 Pruebas diagnósticas en el síndrome hemolítico urémico
|
Síndrome hemolítico urémico
atípico
Pruebas de alteración de la regulación del complemento |
|
Síndrome hemolítico urémico
típico
Muestra fecal si diarrea o frotis rectal: |
|
Púrpura trombocitopénica
trombótica
Actividad plasmática de ADAMTS13 o dosis (ELISA) ± inhibidor MCP: proteína cofactor de membranas; FB: factor B del complemento; THBD: trombomodulina; STEC: E. coli productora de toxina Shiga; Stx: toxina Shiga. |
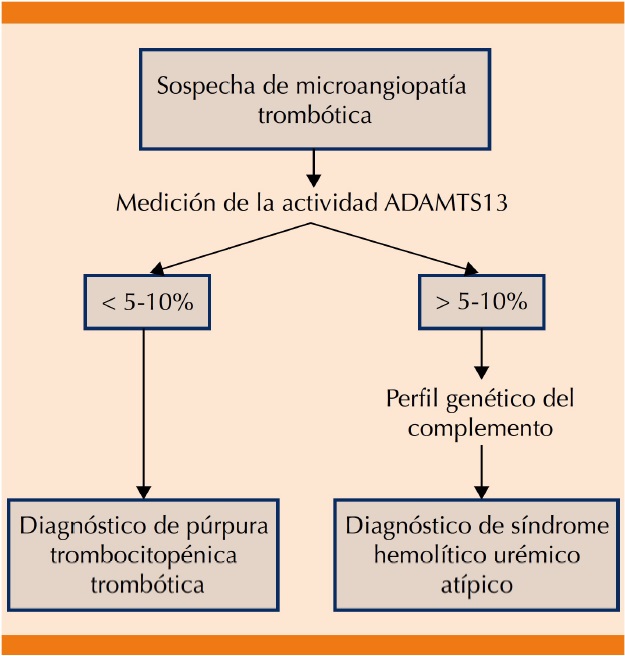
Figura 1 Diagnóstico diferencial: síndrome hemolítico urémico atípico y púrpura trombocitopénica trombótica.
El diagnóstico definitivo de síndrome hemolítico urémico atípico requiere confirmación por pruebas genéticas; sin embargo, se ha encontrado que 40% de los pacientes no muestran alguna alteración genética conocida.30 Asimismo, en la actualidad estas pruebas genéticas no forman parte del diagnóstico inicial del síndrome hemolítico urémico atípico por tres razones: en primer lugar estos estudios genéticos aún no están universalmente disponibles; el síndrome hemolítico urémico atípico sigue siendo un diagnóstico clínico debido a la incapacidad de diagnosticar a todos los pacientes, ya sea bioquímicamente o por genética y, por último, la respuesta al tratamiento no parece estar afectada por la alteración genética.2 Por lo que es necesario una caracterización clínica y paraclínica adecuada del síndrome hemolítico urémico atípico para llegar a un diagnóstico temprano y a un tratamiento oportuno pues hasta ahora muchos consideran el síndrome hemolítico urémico atípico un diagnóstico de exclusión luego de descartar síndrome hemolítico urémico y púrpura trombocitopénica trombótica.
Perfil genético: valor pronóstico
A pesar de que el perfil genético no modifica la respuesta al tratamiento,2 su importancia radica en la influencia que tienen los genes en la aparición de ciertos desenlaces.21 Aunque actualmente las pruebas genéticas son costosas y poco útiles para el diagnóstico inicial, la adecuada caracterización del perfil genético es necesaria debido a la importancia que éstos tienen para el apropiado tratamiento y para repercutir en la morbilidad y mortalidad por esta causa. El Cuadro 2 muestra las principales mutaciones y las características clínicas y paraclínicas relacionadas con éstas.40
Cuadro 2 Características clínicas y paraclínicas de las mutaciones causantes de síndrome hemolítico urémico
| Subgrupo | Frecuencia (%) | Edad de aparición | C3 | Enfermedad renal en etapa terminal (%) | Recurrencia (%) | Recurrencia postransplante (%) |
|---|---|---|---|---|---|---|
| Factor H complemento | 20-30 | < 2 años | N/↓ | 50-70 | 50 | 75-90 |
| Factor I complemento | 4-10 | < 2 años | N/↓ | 50-60 | 10-30 | 45-80 |
| Proteína cofactor de membrana | 5-15 | > 1 año | N/↓ | < 20 | 70-90 | < 20 |
| C3 | 2-10 | Cualquiera | ↓ | 60-80 | 50 | 40-70 |
| Factor B complemento | 1-4 | 1 mes | ↓ | 50-70 | Sí | 100 |
| Molécula inhibitoria trombomodulina | 3-5 | 6 meses | N/↓ | 50-60 | 30 | 1pte |
| Anti-factor H complemento | 6 | 5-13 años | N/↓ | 30-40 | 10-60 | Sí |
La mutaciones en el gen del factor H del complemento son las más comúnmente reportadas y representan alrededor de 30% de los casos;20 asimismo, los autoanticuerpos contra el factor H están presentes en 6 a 10% de los pacientes con síndrome hemolítico urémico atípico3 y esta mutación se relaciona con respuesta incompleta a la terapia plasmática y progresión a enfermedad renal terminal.20 Por otro lado, luego de un análisis, la mutación del factor H junto con la mutación del factor CFI tienen mayor tasa de incidencia de enfermedad renal terminal y muerte durante el primer episodio.27
Los pacientes con mutaciones del factor H tienen riesgo de recurrencia de síndrome hemolítico urémico atípico de 60 a 80% después de un trasplante de riñón41 porque esta intervención no corrige el defecto genético en los pacientes debido a que el factor H es producido principalmente por el hígado, lo que explica el riesgo tan elevado de recurrencia postrasplante; aunque actualmente esta recurrencia ha disminuido debido a la administración profiláctica del eculizumab;42 se han reportado pacientes que después del trasplante renal y a la administración de aculizumab requieren trasplante hepático para anular la activación del complemento.20
Otras alteraciones que pueden sobrevenir es la mutación del CD46 o proteína cofactor de membrana (MCP), una glicoproteína de la superficie celular que protege a las células de la lisis mediada por el complemento,43 si bien en los pacientes con síndrome hemolítico urémico atípico concomitante con deficiencia de CD46 se han reportado recaídas, no respuesta al tratamiento con plasmaféresis y un bajo riesgo de progresión a insuficiencia renal terminal.43
Las mutaciones en el complemento 3 (C3) causan activación incontrolable del C3 convertasa, las concentraciones de C3 durante el cuadro disminuyen y la mutación se asocia con mal pronóstico porque entre 60 y 80% progresan a enfermedad renal terminal y la tasa de recurrencia del síndrome luego del trasplante renal es muy alta.44
Las mutaciones del factor B (CFB) son extremadamente raras, sólo se han reportado algunos casos y el reporte de éstos es esporádico;45 muchos de estos pacientes mueren de manera temprana, por tal motivo, la frecuencia y recurrencia aún no están bien establecidas.
Aunque muchos estudios claramente relacionan las mutaciones de los genes del complemento con la patogénesis del síndrome hemolítico urémico atípico, es probable que existan otras alteraciones genéticas adicionales a esta enfermedad, y actualmente se plantean tres evidencias que lo soportan. En primer lugar, en un gran porcentaje de casos, no se encuentran mutaciones en los genes del complemento comúnmente implicados.8 Segundo, existe una tasa extremadamente alta de penetrancia incompleta en el síndrome hemolítico urémico atípico familiar, lo que es consistente con la existencia de factores genéticos adicionales.8 Tercero, los estudios actuales se han enfocado en menos de una docena de genes,8 aunque el sistema de complemento es un sistema grande e integrado con muchas otras vías y componentes.
De acuerdo con lo anterior, con los nuevos descubrimientos, como las mutaciones del gen DGKE que codifica la diacilglicerol cinasa-ε,7-46 el concepto que tenemos de la enfermedad puede ir cambiando porque esta molécula no tiene relación alguna con la regulación del complemento, por el contrario, forma parte de la vía de la coagulación y su deficiencia se manifiesta como una microangiopatía trombótica.
CONCLUSIONES
El síndrome hemolítico urémico atípico es una afección poco frecuente, pero sin un diagnóstico temprano y un tratamiento oportuno se asocia con complicaciones importantes. Es importante identificar qué pacientes con microangiopatía trombótica tienen riesgo de este padecimiento y son aptos para la realización de un perfil genético porque éste es la única herramienta diagnóstica que permite diferenciar por completo el síndrome hemolítico urémico atípico de otras causas de microangiopatía trombótica.

