











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkANTECEDENTES
La enfermedad de Kikuchi-Fujimoto, o linfadenitis necrosante histiocítica benigna, es una entidad clínica que fue descrita en Japón en 1972 por Kikuchi y de manera casi simultánea ese mismo año también fue descrita por Fujimoto de manera independiente. La describieron como una linfadenitis con proliferación focal de células reticulares acompañadas de numerosos histiocitos y residuos nucleares extensos.1 La enfermedad de Kikuchi-Fujimoto generalmente es de alivio espontáneo y tiene buen pronóstico porque tiene baja tasa de complicaciones, aunque existen casos reportados en los que sobrevino como manifestación de exacerbación de enfermedades del tejido conectivo y tuvieron desenlaces fatales.2,3
La enfermedad de Kikuchi-Fujimoto es un padecimiento poco frecuente, cuya incidencia real sigue siendo desconocida. Se han reportado casos en todo el mundo en todos los grupos étnicos, predomina en la población japonesa y otros grupos étnicos de origen asiático. Por lo general, afecta a pacientes menores de 30 años, pero existen casos documentados desde 19 meses hasta 75 años. Antes se creía que la enfermedad de Kikuchi-Fujimoto era cuatro veces más frecuente en mujeres que en hombres, pero estudios más recientes señalan que podría afectar a mujeres y hombres de manera similar.4,5
La causa exacta de la enfermedad de Kikuchi-Fujimoto sigue siendo desconocida. Puede ser un padecimiento mediado por autoinmunidad o una respuesta inmunitaria exagerada a algún agente infeccioso subyacente. Las características clínicas e histológicas de la enfermedad sugieren un origen viral, el virus de Epstein-Barr es el que se asocia de manera más frecuente. 5 La enfermedad de Kikuchi-Fujimoto se relaciona frecuentemente con otros trastornos autoinmunitarios, el lupus eritematoso sistémico es el más comúnmente asociado.2 En muchos reportes, la enfermedad de Kikuchi-Fujimoto precedía a la aparición de lupus eritematoso sistémico; aunque existen reportes en los que se diagnosticó de manera simultánea o después del diagnóstico de lupus eritematoso sistémico. Existen hallazgos clínicos compatibles con esta enfermedad y que no es frecuente encontrar en enfermedad de Kikuchi-Fujimoto, como organomegalias y existencia de anticuerpos antinucleares.6
La enfermedad de Kikuchi-Fujimoto se manifiesta como enfermedad aguda o subaguda con síntomas B y linfadenopatía cervical posterior dolorosa. Los ganglios linfáticos son pequeños, usualmente de 0.5 a 4 cm. En 2006 Kucukardali y colaboradores analizaron 244 casos de enfermedad de Kikuchi-Fujimoto, la fiebre fue el síntoma sistémico más frecuente (afectó a 35% de los pacientes). La linfadenopatía se encontró en el 100% de los pacientes, exantemas eritematosos en 10% y hepatoesplenomegalia en 3%. La asociación con lupus eritematoso sistémico estuvo presente en 13% de los pacientes y con infecciones virales en 10%. El órgano extranodal afectado con más frecuencia es la piel que, de acuerdo con otros estudios, puede afectarse hasta en 30% de los pacientes con enfermedad de Kikuchi-Fujimoto, las manifestaciones fueron pápulas, eritema malar, placas o nódulos.4,6 En los análisis de laboratorio se ha descrito de manera frecuente leucopenia y anemia; otros hallazgos son elevación de la velocidad de sedimentación globular, deshidrogenasa láctica y de la alanina aminotransferasa.1,6
El diagnóstico de la enfermedad de Kikuchi-Fujimoto se establece de manera general con base en una biopsia por escisión de los ganglios linfáticos afectados. En términos histológicos, en los ganglios linfáticos se distinguen áreas focales de necrosis en parches, con infiltrados de histiocitos, dendrocitos plasmocitoides e inmunoblastos mezclados con restos celulares y, de manera característica, ausencia de neutrófilos.1,7 El diagnóstico diferencial de la enfermedad de Kikuchi-Fujimoto es amplio y deben excluirse infecciones como tuberculosis, toxoplasmosis, por Bartonella henselae, VIH y virus de Epstein-Barr; también enfermedades del tejido conectivo (como lupus eritematoso sistémico) y padecimientos linfoproliferativos.6
No existen guías de tratamiento contra la enfermedad de Kikuchi-Fujimoto y las recomendaciones están basadas en los reportes de caso y opinión de expertos. Debido a su naturaleza autolimitada, la observación es el abordaje más frecuente. Los pacientes sintomáticos o con tejidos extranodales implicados, como el sistema nervioso central, la piel y los ojos pueden beneficiarse con pulsos de corticoesteroides, antiinflamatorios no esteroides y antipiréticos. Otros fármacos que se han estudiado para el tratamiento de esta entidad han sido los antimaláricos con resultados variables.6
La siliconosis es un tipo de enfermedad autoinmunitaria con manifestaciones sistémicas inespecíficas. Se agrupa en el conjunto de enfermedades que pueden manifestarse como síndrome autoinmunitario inducido por coadyuvantes. 8 El silicón es un polímero de dimetilsiloxano usado en el ámbito médico por su viscosidad variable según la longitud del polímero. En décadas recientes millones de personas se han expuesto al silicón de distintas maneras, desde objetos de uso diario en el hogar, como mamilas, a productos médicos implantables como válvulas cardiacas o derivaciones ventrículo-peritoneales, pero la forma más popular del uso de silicón es para implantes mamarios, que después de décadas de investigación es considerado el material ideal para la mamoplastia de aumento.9
Se ha descrito la relación del uso del silicón con enfermedades autoinmunitarias, que incluyen artritis reumatoide, lupus eritematoso sistémico, polimiositis, enfermedad mixta del tejido conectivo, síndrome de Sjögren y esclerosis sistémica.9 Una explicación por la que el silicón produce respuesta inflamatoria es porque puede actuar como coadyuvante inmunogénico provocando una respuesta local y sistémica Th1 y Th17. De manera alternativa, el silicón puede inducir una respuesta por reacción cruzada con el tejido conectivo que contiene glucosaminoglicanos, que es una molécula estructuralmente similar al silicón y se encuentra en el tejido conectivo.8,9 Se ha demostrado que los pacientes que padecen este síndrome tienen mayor prevalencia de los alelos HLA-DQ2 y DRW53, lo que indica una predisposición genética.8,10
El diagnóstico es complicado y por exclusión. La historia clínica y los antecedentes sirven para la sospecha, no existe un estudio sérico que sea específico para este padecimiento. Asimismo, algunos estudios de imagen demostraron ser de utilidad para la localización del silicón a distancia y en el estudio histopatológico puede haber hallazgos compatibles con siliconosis.8 En 2015 Alijotas-Reig propuso nuevos criterios diagnósticos de síndrome autoinmunitario inducido por coadyuvantes con base sólo en datos clínicos y de laboratorio objetivos, pero no han sido validados (Cuadro 1).11
Cuadro 1 Criterios diagnósticos de enfermedad por coadyuvantes humana (ASIA) propuestos por Alijotas-Reig
Tomado de la referencia 11.
El tratamiento del síndrome autoinmunitario inducido por coadyuvantes se basa en la eliminación del estímulo externo y en la mayoría de los casos se observa una respuesta favorable a largo plazo sin necesidad de iniciar tratamiento inmunomodulador. Sin embargo, en los casos de evolución a enfermedades autoinmunitarias/autoinflamatorias, por lo general es necesario iniciar tratamiento inmunomodulador.8
CASO CLÍNICO
Paciente femenina de 31 años de edad, casada, empleada de oficina. Consumo de alcohol y tabaquismo negados. Con antecedente de un episodio de depresión mayor diagnosticado 12 años previos y colocación de implantes mamarios 10 años antes. Negó antecedentes alérgicos, traumáticos o transfusionales.
La paciente padeció un cuadro de seis días de evolución caracterizado por cefalea de tipo punzante holocraneana de predominio biparietal de intensidad 7/10, irradiada al cuello, acompañada de parestesias faciales, por lo que se automedicó analgésicos no especificados, con lo que los síntomas remitieron parcialmente. Doce horas posteriores al inicio de los síntomas se acompañó de ataque al estado general y fiebre no cuantificada, náusea y vómito de contenido gastroalimentario en 10 ocasiones y 15 evacuaciones líquidas sin moco ni sangre, por lo que acudió con un médico particular quien indicó tratamiento antibiótico con ciprofloxacino y antiinflamatorios. Por falta de mejoría acudió al servicio de urgencias a valoración por persistir con fiebre cuantificada hasta 38.5°C, cefalea de características similares a las comentadas, además de fotofobia y fonofobia. Al interrogatorio dirigido negó fosfenos, acúfenos, dolor abdominal, faringodinia, tos, disnea, dolor torácico, síntomas urinarios, pérdida de peso reciente o viajes recientes. A la exploración física inicial se encontró con presión arterial de 90/70 mmHg, frecuencia cardiaca de 75 latidos por minuto, eutérmica. Dolor a la palpación media y profunda del abdomen de manera generalizada, sin datos de irritación peritoneal ni organomegalias palpables. Neurológicamente íntegra sin datos de irritación meníngea. En los estudios paraclínicos de laboratorio se encontró hipocalemia leve 3.2 mmol (3.5-5.5), leucopenia 2000 U/mL (4.8-10.8), trombocitopenia 120,000 U/mL (150,000-400,000) y bandemia 35%; además de deshidrogenasa láctica de 387 UI/L. Examen general de orina sin datos patológicos. Radiografía de tórax sin alteraciones y radiografía de abdomen con escasos niveles hidroaéreos en el cuadrante inferior derecho.
Se decidió su internamiento con diagnóstico de gastroenteritis aguda infecciosa para continuar con tratamiento antibiótico, corrección de la hipocalemia y vigilancia. Al tercer día de estancia intrahospitalaria tuvo remisión de los síntomas gastrointestinales, pero persistió con cefalea moderada a intensa y fiebre intermitente cuantificada hasta 39.0°C sin predominio de horario, por lo que se cambió el esquema antibiótico a ceftriaxona.
Durante su internamiento las concentraciones de hemoglobina fueron disminuyendo hasta padecer anemia leve normocítica normocrómica de 10.8 g/dL (12-16).
Una semana después de su internamiento la paciente continuó con fiebre, por lo que se realizó protocolo de fiebre de origen desconocido. Se realizó tomografía toracoabdominal, examen general de orina, urocultivo, pruebas de funcionamiento hepático, deshidrogenasa láctica, hemocultivos, anticuerpos antinucleares, factor reumatoide, anticuerpos para virus de hepatitis, PCR para virus de Epstein-Barr, citomegalovirus, VIH, parvovirus, Brucella; anticuerpos antinucleares, factor reumatoide, anticoagulante lúpico, anticardiolipina, anti B2 GP1, anticuerpos anti-Ro, La, RNP, Sm, Scl-70 y Jo1, sólo se detectó persistencia en la elevación de deshidrogenasa láctica (462 U/L). El resto de los estudios de laboratorio no mostró hallazgos relevantes. En la tomografía de tórax se encontraron adenomegalias cervicales y axilares bilaterales con predominio derecho a nivel supraclavicular y axilar con infiltración de los planos grasos vecinos, por lo que se decidió intervención quirúrgica para resección de conglomerado ganglionar y análisis histopatológico. Se llevó a cabo la intervención quirúrgica sin incidentes ni accidentes con resección de cúmulo ganglionar con reporte histopatológico de linfadenitis histiocítica necrosante sobreimpuesta en linfadenopatía por silicón descartando la posibilidad de proceso linfoproliferativo (Figura 1), se practicó inmunohistoquímica con anticuerpos primarios positivos para CD20, CD3, Ki67 con patrón reactivo; MPO y CD68 positivo en macrófagos; CD30 positivo en inmunoblastos y negativos para LMP.
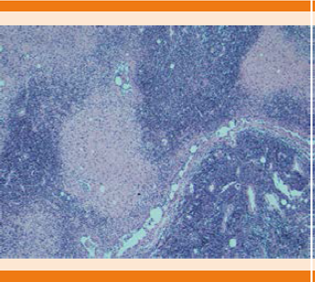
Figura 1 Estudio histológico que muestra infiltrado de histiocitos con áreas focales de necrosis sobre linfadenopatía por silicón (tinción con hematoxilina y eosina; amplificación original x 20).
Con base en este hallazgo se dio tratamiento sintomático y durante la tercera semana de su estancia intrahospitalaria se realizó intervención quirúrgica para retiro de implantes mamarios de silicón, con posterior alivio de los síntomas, permaneciendo afebril hasta la remisión total de los síntomas, por lo que se decidió su egreso para seguimiento ambulatorio.
Se envió material de prótesis a estudio histopatólógico que reportó cápsulas de glándula mamaria derecha e izquierda con tejido conectivo denso fibroso con inflamación crónica xantomatosa inespescífica y seis ganglios linfáticos axilares con inflamación crónica xantogranulomatosa a silicón con necrosis focal residual.
DISCUSIÓN
No existen casos documentados de enfermedad de Kikuchi-Fujimoto secundaria a siliconosis. La fisiopatología de la enfermedad de Kikuchi-Fujimoto tiene un sustento autoinmunitario y la siliconosis provoca una respuesta de tipo autoinmunitario en el paciente. Una posibilidad es que la siliconosis haya provocado la enfermedad de Kikuchi-Fujimoto, aunque no hay reportes que hablen de coadyuvantes como probables desencadenantes de este tipo de linfadenopatía.
La enfermedad de Kikuchi-Fujimoto tuvo una manifestación habitual en la paciente con fiebre y adenopatías cervicales, quien no tuvo manifestaciones dermatológicas, que también son frecuentes en estos pacientes. La paciente tenía cuatro criterios mayores y uno menor para diagnóstico del síndrome autoinmunitario inducido por coadyuvantes según los nuevos criterios propuestos en 2015, y posterior al retiro del material protésico se ha mantenido asintomática. En este caso el diagnóstico se estableció gracias a la biopsia de ganglio linfático.
No existen reportes de pacientes que hayan mostrado una relación entre estas dos afecciones clínicas. Es necesario un mejor reporte epidemiológico para poder establecer a la siliconosis como una causa de enfermedad de Kikuchi-Fujimoto y para conocer más a fondo la relación entre estos padecimientos.

