











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkANTECEDENTES
El escrito en cuestión se centra en el estudio de la agregometría plaquetaria, que sería imposible entender por el clínico si no se tiene el adecuado conocimiento de la fisiología plaquetaria, de igual manera, este artículo revisa los trastornos identificables con esta prueba diagnóstica.
La función de las plaquetas y su disfunción secundaria a trastornos hereditarios y adquiridos son conceptos importantes que deben conocerse para poder resolverlos en situaciones de urgencia y en el actuar diario del médico. Al paso de los años se han diseñado diversos métodos para evaluar la función plaquetaria, que no son bien entendidos y en el peor de los casos conocidos por la comunidad médica.
Fisiología y morfología plaquetaria
Las plaquetas son células sanguíneas fundamentales para la hemostasia y son las principales implicadas en alteraciones como la trombosis, trastornos hemorrágicos y en eventos trombóticos hereditarios o adquiridos.
El estudio de la función plaquetaria inició hace más de 100 años, cuando al estudiarlas se identificaron características únicas que eran decisivas para la hemostasia y la trombosis;1 a pesar de todos los avances en el conocimiento de la fisiología y la morfología plaquetaria no se ha podido resolver de forma adecuada una cuestión fundamental, y esa cuestión es la capacidad de simular y estudiar la función plaquetaria de cada uno de nuestros pacientes, esto debido a que las plaquetas son sensibles a la manipulación y se activan en los tubos de vidrio. Debido a este problema en 1960 se desarrolló una técnica que resolvería parcialmente este problema: la agregometría plaquetaria. A través de ésta se ha logrado entender a fondo la fisiología de la hemostasia y su interpretación en las alteraciones de la misma.2
Origen y estructura plaquetaria
Las plaquetas son células anucleadas con forma discoide de aproximadamente 0.5 x 3.0 µm, tienen su origen de los megacariocitos a través de un proceso endomitótico.3 La trombopoyetina es la hormona que permite el adecuado desarrollo de las plaquetas que, a diferencia de la eritropoyetina, se sintetiza en el músculo liso y la médula ósea y no exclusivamente en el riñón o en el hígado, y se elimina a través de las mismas plaquetas que forma, por tanto, a mayor destrucción plaquetaria, mayor concentración de trombopoyetina circulante.4
Al tener en mente que las plaquetas son células, debemos tener en consideración que los principales organelos contenidos en ella son mitocondrias, lisosomas, peroxisomas, gránulos (cuerpos) alfa y gránulos densos. Estos dos últimos son especialmente importantes porque tienen una gran cantidad de factores que influyen en la coagulación. Los gránulos alfa contienen selectina P, factor V, factor VIII, factor de von Willebrand, trombospondina, fibronectina, fibrinógeno, β-tromboglobulina, factor plaquetario 4 y factor de crecimiento derivado de plaquetas (PDGF por sus siglas en inglés). Los gránulos densos almacenan adenosín difosfato (ADP), calcio y serotonina, a su vez, el citoplasma puede contener otras sustancias, como: serotonina, epinefrina, norepinefrina, óxido nítrico y citocinas.5
Las plaquetas participan en la hemostasia y la trombosis, esto lo consiguen adhiriéndose al endotelio vascular dañado. Las plaquetas interactúan con factores ambientales y con otras plaquetas, creando procesos complejos que se originan en la superficie de la membrana plaquetaria, esta membrana, a su vez, proporciona una interfase reactiva entre las plaquetas y el exterior utilizando los receptores localizados en la superficie. Estos receptores son primordiales para la transducción de señales y estímulos externos hacia el interior de la misma.6 La plaquetas se activan con la interacción que se origina entre los diferentes receptores de membrana y un gran número de moléculas pequeñas, enzimas y complejos proteicos macromoleculares que tienen la finalidad de contraer el citoesqueleto de la misma.
Otra característica impresionante de estas células es que pueden cambiar de forma, transformándose de una célula discoide a una esférica, con la finalidad de tener extensiones (pseudópodos) que faciliten la adhesión al endotelio y otras células, así como la interacción con otras plaquetas y liberación del contenido de los gránulos en su interior.6 La activación plaquetaria depende de múltiples estímulos que tienen como finalidad generar una secuencia de eventos. Estos estímulos son: trombina, tripsina, colágena, ADP, epinefrina, metabolitos del ácido araquidónico, factor activador de plaquetas y epinefrina (Figura 1).
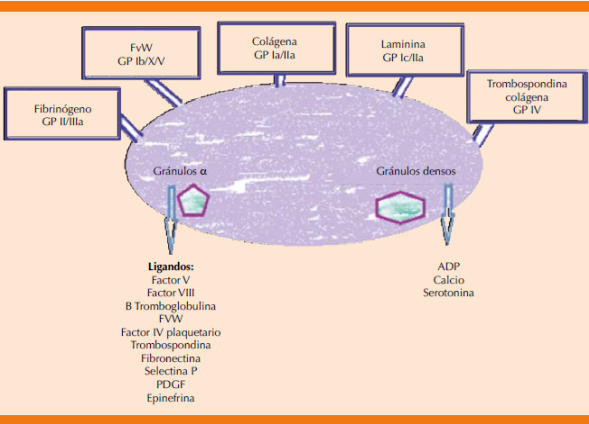
La activación depende de la interacción entre el medio externo e interno, se consigue a través de receptores de membrana, a continuación se enumeran los principales receptores de la membrana plaquetaria:
Receptores de ADP. El adenosín difosfato (ADP) juega un papel central en la hemostasia al actuar como cofactor esencial en la activación plaquetaria. El ADP está presente en altas concentraciones en los gránulos densos y se libera a través de la activación plaquetaria ejerciendo un reforzamiento para la agregación de la misma.7
Receptores de epinefrina. La estimulación con epinefrina se da a través de la estimulación de los receptores α2-adrenérgicos, de éstos se expresan aproximadamente 300 receptores en cada plaqueta. Una cualidad peculiar de la epinefrina es que activa la agregación plaquetaria sin alterar la morfología de la misma.6
Receptores de serotonina. La serotonina (5-hidroxitriptamina, 5-HT) estimula y activa las plaquetas, mismas que, una vez activadas, aumentan la liberación de serotonina mediante los gránulos densos, lo que aumenta la respuesta plaquetaria a través de receptores específicos 5-HT2A.8
Receptores de tromboxano A2. La fosfolipasa A2 se activa a través de agonistas de la activación plaquetaria, ocasionando que el ácido araquidónico se separe de los fosfolípidos (localizados en la membrana), el mismo, a su vez se metaboliza a sustancias plaquetarias activas, como los tromboxanos (TX) y su precursor PGH2. Es importante conocer toda esta vía metabólica porque los tromboxanos estimulan el cambio de forma, la secreción y la agregación plaquetaria. El tromboxano A2 es el más específico y potente. 9
Receptores de vasopresina. La vasopresina interactúa con las plaquetas para inducir cambios de forma, secreción de los gránulos y agregación.
Receptor del factor activador de plaquetas. Es un agonista único porque es un fosfolípido que estimula las plaquetas en concentraciones nanomolares. Este agonista es sintetizado por varias células inflamatorias y las mismas plaquetas causan inducción de cambios de forma, secreción y agregación.10
Receptores de trombina. La trombina es un potente activador plaquetario.
Receptores de colágena. La colágena induce adhesión y activación plaquetaria a través de varias proteínas de membrana, como GP Ia-IIa, GP IV, GP VI y GP V.11
Receptores para la adhesión plaquetaria. Las glucoproteínas (GP) de la superficie de la membrana participan de forma crítica en la adhesión de las plaquetas al subendotelio expuesto (colágena, fibronectina y factor de von Willebrand) e interactuando con otras plaquetas y componentes de la sangre (fibrinógeno y factor de von Willebrand). Se enumeran de mayor a menor tamaño de la I a la IX.12
Estas glucoproteínas pertenecen a la familia de las integrinas, son complejos proteicos heterodiméricos α/β.13 La glucoproteína más abundante es la IIb/IIIa (αIIb/β3), esta glucoproteína es responsable de la unión plaquetaria al fibrinógeno, colágena, protrombina, factor de von Willebrand, trombospondina y vitronectina, gracias a estos procesos de unión es el principal promotor de la agregación plaquetaria.14
El complejo GP Ib-IX-V es el segundo receptor plaquetario más abundante y participa principalmente en la adhesión plaquetaria al subendotelio mediante la unión al factor de von Willebrand (FvW).15 También promueve la unión a la trombina y facilita la activación de otras plaquetas.
Función plaquetaria
La hemostasia primaria es el proceso inicial de la coagulación y tiene el objeto de crear un tapón plaquetario en respuesta a daño al endotelio vascular. La hemostasia primaria consiste de tres fases: adhesión, activación, secreción y agregación plaquetaria. En condiciones normales, las plaquetas no tienen contacto con la matriz de tejido conectivo del subendotelio vascular. Cuando se rompe la integridad endotelial, se exponen fibras de colágena, factor de von Willebrand y otras proteínas de la matriz subendotelial,16 y es la interacción de las plaquetas con estas sustancias lo que proporciona la superficie para la adhesión plaquetaria y sirve como un fuerte estímulo para la activación de las plaquetas. En condiciones de gran fricción por flujo sanguíneo (arterias y capilares), esta interacción consiste de manera primordial en la unión de la GP Ib al factor de von Willebrand, que se adhiere a la colágena subendotelial.17,18 En condiciones de baja fricción, la GP Ia/IIa puede unirse de manera directa a la colágena.
La unión de los receptores de superficie de las plaquetas con sus ligandos activa varias reacciones bioquímicas de señalización intracelular a través de segundos mensajeros, como las tirosinacinasas, el calcio, la fosfolipasa C, el fosfoinositol 3 cinasa y el AMP cíclico, entre otros.19
Esta señalización produce cuatro cambios mayores en las plaquetas:
Se produce un rearreglo del citoesqueleto de actina que causa aplanamiento de las plaquetas y extensión de pseudópodos (filópodos y lamelópodos) para sellar el defecto endotelial.20
La activación de la fosfolipasa A2 libera ácido araquidónico de los fosfolípidos de la membrana y se convierte en prostaglandinas y tromboxano A2 (TXA),9 aumentando la vasoconstricción e induciendo taponamiento local.
Los gránulos intracelulares se unen con el sistema canalicular abierto de la membrana y liberan su contenido en el plasma circundante. Los gránulos densos liberan ADP y serotonina, entre otras sustancias, que interactúan con los receptores celulares de superficie de otras plaquetas, amplificando la activación y estimulando la agregación.16,21
La activación plaquetaria incrementa la concentración de GP IIb/IIIa en la membrana e induce cambios en su conformación, lo que permite su unión a fibrinógeno soluble y agregación de las plaquetas.14
La trombina y la epinefrina son de igual forma fundamentales para la activación y agregación plaquetaria, actuando a través de los receptores mencionados.
La agregación plaquetaria se regula de manera primaria por la unión de GP IIb/IIIa a fibrinógeno y en menor medida a factor de von Willebrand y fibronectina.22 Estas moléculas, al ser polivalentes, fungen como un puente de unión entre varias plaquetas a la vez. Esta unión plaqueta-fibrinógeno-plaqueta activa el proceso de agregación del coágulo de plaquetas. Una vez formado el coágulo plaquetario, debe estabilizarse mediante el desarrollo de una red circundante de fibrina, que es formada por los factores de coagulación, que se activan simultáneamente con las plaquetas.
Las plaquetas y los factores de coagulación no sólo se activan de manera simultánea y paralela, sino que se ha observado que las plaquetas son decisivas para activar y regular la cascada de coagulación, porque la membrana activada de las plaquetas contiene fosfatidilserina, éste es un fosfolípido que se encuentra en concentraciones altas y funciona como un cofactor para la unión y activación de los factores de coagulación. Asimismo, las plaquetas contienen en sus gránulos alfa factores de coagulación y calcio en los gránulos densos. Por último, el factor Va unido a plaquetas se mantiene en relativa “protección” para no ser desactivado por el complejo proteína C/S.16,23
Agregometría plaquetaria
La historia de la agregometría plaquetaria inicia en 1962, cuando fue descrita por Born como una técnica que estimaba la cinética de la agregación de las plaquetas a través de la turbidometría (midiendo la turbidez o densidad óptica del plasma). Posterior a su descubrimiento inmediatamente se volvió el patrón de referencia para evaluar la función plaquetaria.24
A continuación se describe la metodología para desarrollar una agregometría convencional y los diferentes métodos que se han desarrollado para evaluar la agregación plaquetaria desde 1960.
En la agregometría convencional inicialmente se centrifuga la sangre a baja velocidad (1000 rpm) durante 15 minutos para obtener un plasma rico en plaquetas, este plasma se coloca en un recipiente a temperatura de 37°C entre una fuente de luz y una celda solar que va a sensar la luminiscencia del plasma. También se requiere la preparación de una muestra de plasma pobre en plaquetas (PPP), que se obtiene por centrifugación rápida (3000 rpm). La calibración de la trasmisión de la luz a través del PPP (plasma claro) se ajusta al 100%; la transmisión de luz a través del PRP (plasma rico en plaquetas, plasma turbio u opaco) entonces se ajusta a 0% (Figura 2).25
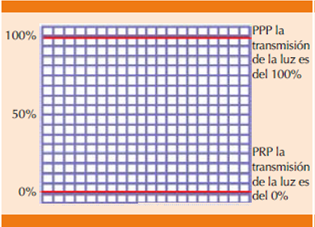
Figura 2 Calibración y gráfica de la transmisión de la luz con plasma rico en plaquetas (PRP) y pobre en plaquetas (PPP).
Una vez que se tiene el plasma colocado de la forma mencionada debe agregarse un agonista plaquetario, como epinefrina, ADP, colágena, ácido araquidónico, trombina y ristocetina (un antibiótico que estimula la agregación por aumento de la unión del factor de von Willebrand a la GP Ib), con el que se agregarán las plaquetas, lo que resulta en el aumento de la trasmisión de la luz (disminución de la turbidez del plasma) que se detecta y registra como una función de tiempo posterior a la adición del agonista. A mayor agregación plaquetaria, mayor trasmisión de la luz y viceversa (Figura 3).25 Estos cambios de luminiscencia o turbidez son el reflejo de la formación de agregados plaquetarios relativamente grandes, derivados de microagregados.

Figura 3 Esquema de la agregación plaquetaria en plasma rico en plaquetas. En la figura superior se ejemplifica la falta de transmisión de la luz a través del plasma turbio. En la figura inferior se induce agregación plaquetaria con la adición de un agonista (epinefrina); por eso se agregan las plaquetas y permiten el paso de la luz y se grafica en porcentaje.
El uso de agonistas (ADP, colágeno, epinefrina, ristocetina) en un rango de concentraciones diferentes resulta en respuesta plaquetaria clásica, incluido el cambio en la forma de los agregados, así como la agregación primaria y secundaria.
La existencia de inhibidores de función plaquetaria, así como la concentración del agonista facilita la detección de trastornos plaquetarios clásicos, basados en el patrón de agregación (Cuadro 1).104
Cuadro 1 Patrones de agregación plaquetaria en alteraciones específicas de la función plaquetaria
| Enfermedad | ADP primaria | ADP secundaria | Colágena | Ristocetina | Otras consideraciones |
|---|---|---|---|---|---|
| Enfermedad de von Willebrand | ++++ | ++++ | ++++ | Muy variable | Morfología normal de las plaquetas, el panel de enfermedad de von Willebrand es usualmente anormal |
| Síndrome de Bernard-Soulier | ++++ | ++++ | ++++ | 0 | Plaquetas gigantes. Panel de trombocitopenia- enfermedad de von Willebrand normal |
| Trombastenia de Glanzmann | 0 | 0 | 0 | +++ | Morfología plaquetaria normal |
| Enfermedad de la poza de almacenamiento | ++++ | 0 a ++ | ++++ | ++++ | Morfología plaquetaria normal (excepto en el grupo de plaqueta gris), microscopia electrónica anormal |
| Defecto de secreción | ++++ | 0 a ++ | ++++ | ++++ | Morfología normal con microscopia electrónica y óptica |
++++: respuesta normal; +++: respuesta ligeramente disminuida; ++: respuesta disminuida; +: respuesta muy disminuida; 0: sin respuesta; ADP: adenosín difosfato.
Su graficación genera una curva de la agregación plaquetaria en porcentaje y tiempo. La curva típica de la agregación plaquetaria tiene cinco fases distintas con dos curvas (Figura 4).25,26
Incremento inicial de la transmisión de la luz atribuible a dilución del PRP por el agonista que añade.
Leve disminución de la transmisión a medida que las plaquetas cambian de forma.
Incremento agudo inicial de la transmisión a medida que las plaquetas se agregan (agregación primaria).
Meseta breve o disminución de la agregación.
Agregación secundaria que se estimula por la secreción de los gránulos plaquetarios.
En la mayoría de los individuos esta respuesta bifásica se observa con concentraciones bajas de ADP y epinefrina.

1: agregación primaria; 2: meseta; 3: agregación secundaria.
Figura 4 Curva típica de la agregación plaquetaria en PRP con epinefrina o ADP. La flecha indica la dilución del PRP al agregar el agobiata.
Las curvas que se generan con otros agonistas (colágena, ristocetina) son típicamente unimodales con una sola curva (Figura 5). Este método tiene varias limitaciones inherentes; consume tiempo, es laborioso y complejo, por lo que se requiere un equipo humano bien adiestrado y con experiencia en la realización de la técnica.

La muestra debe obtenerse por punción venosa directa y se colecta en tubos con anticoagulante a base de citrato (tubos para tiempos de coagulación con tapón de color lila). La prueba debe realizarse en las tres horas después de obtener la muestra. La centrifugación puede dañar o activar las plaquetas y no debe realizarse en muestras hemolizadas, lipémicas o con contaminación por eritrocitos.27-29 Al igual que en otros estudios de funcionalidad plaquetaria, la agregometría puede verse alterada por las concentraciones de plaquetas en cada paciente, la trombocitopenia menor de 100 x 109/L o la trombocitosis mayor de 400 x 109/L pueden alterar los resultados y debe tenerse en cuenta para su interpretación.
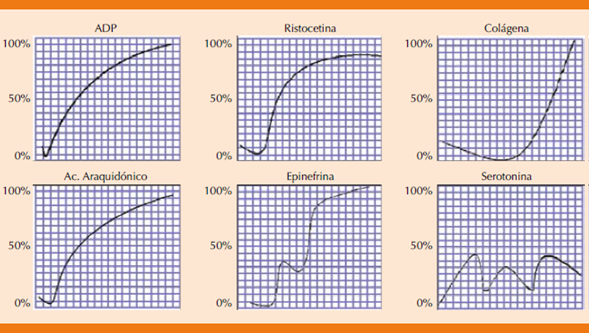
Las dosis de los agentes agregantes pueden variar en los diferentes centros. Las dosis estandarizadas son las siguientes: la epinefrina, por lo regular, se utiliza en dos dosis, la dosis alta en concentración de 2.5 x 10-5 y la dosis baja en concentración de 2.5 x 10-6. De manera similar, el ADP también se utiliza en dos concentraciones, la dosis alta en concentración de 2.0 x 10-5 y la dosis baja de 2.5 x 10-6; la colágena se utiliza en concentración final de 0.19 mg/mL; ristocetina en concentración de 1.25 mg/mL y ácido araquidónico en concentración de 0.5 mg/mL.30 Los valores normales de la agregación plaquetaria con los diferentes agonistas se describen en el (Cuadro 2) y los patrones normales de las curvas con cada agonista de manera individual se muestran en la (Figura 6).
Cuadro 2 Valores normales de agregación plaquetaria según cada agonista
| Agonista | Concentración | Agregación (%) |
|---|---|---|
| Adenosín difosfato | 5 µm 10 µm | 68-88 71-88 |
| Ácido araquidónico | 0.5 mm | 74-99 |
| Colágena | 2 µg/mL | 70-94 |
| Epinefrina | 5 µm | 78-88 |
| Ristocetina | 1.25 mg/mL | 87-102 |
En la actualidad hay agregómetros computados modernos con capacidad multicanal, que tienen la capacidad de medir de manera más precisa la luminiscencia de los agregados al ADP u otros agonistas. A pesar de los avances descritos, existe una gran limitación, la técnica no se ha estandarizado adecuadamente.112-114 Por esta razón la principal deficiencia de la agregometría es que no es lo suficientemente sensible para evaluar la formación de microagregados plaquetarios.115,116
La agregometría es incapaz de detectar agregados menores a 100 plaquetas.117 De igual forma subestima el grado y la duración de inhibición plaquetaria de fármacos, como los anti Gp IIb/IIIa (por ejemplo, abciximab).35
En la actualidad existen otros métodos agregométricos más complejos, como la agregometría de sangre total, los métodos de dispersión de luz y la prueba Verify Now.
La agregometría de sangre total se desarrolló en 1980, por Cardinal y Flower31 en respuesta a las limitaciones de la agregometría estándar.31 El método consiste en mezclar sangre total a 37ºC entre dos electrodos de platino dispuestos a una distancia fija.32 Posteriormente los electrodos se cubren de plaquetas, después se utiliza un agonista que aumenta la adhesión plaquetaria, causando un cambio en el tiempo en la masa plaquetaria adherida a los electrodos, alterando la resistencia al flujo de electricidad entre los electrodos, este cambio se registra con impedancia y se monitorea con una grabadora. El aumento en la resistencia al flujo de electricidad es proporcional a la agregación de las plaquetas en torno a los electrodos y se genera una curva de agregación.33
A pesar de lo que se esperaba, este método da resultados similares a la agregometría convencional, es insensible para detectar agregados pequeños, pero es superior cuando se utiliza para vigilar el tratamiento con antiagregantes plaquetarios.34,35
Se ha observado que este método es especialmente útil para diagnosticar y dar seguimiento de defectos plaquetarios, estados de hiperactividad plaquetaria (por ejemplo, síndrome de la plaqueta pegajosa) y vigilar el efecto farmacológico de aspirina y clopidogrel.34-37,105
Los métodos de dispersión de luz se basaron inicialmente en la citometría de flujo. Ésta es muy efectiva para contar plaquetas y monitorear la formación de agregados, sin importar el tamaño de los mismos, durante la fase de agregación temprana,106 pero era imposible evaluar los mismos en fases tardías. Por esta razón en los últimos años se desarrolló un agregómetro (PA-200, Kowa Ltd.) que combina el uso de dispersión de luz con láser y la agregometría convencional, monitoreando la formación de microagregados de hasta dos o tres plaquetas.107,108
La metodología de este agregómetro se basa en la observación de que la intensidad de la dispersión lumínica es proporcional al tamaño de los agregados, sensando no sólo el tamaño, sino también el número de agregados. Otra gran ventaja es que usa concentraciones de agonista más fisiológicas, concentraciones hasta tres veces menores que el método convencional.
Esta tecnología puede ser útil para detectar hiperreactividad plaquetaria en varias enfermedades (síndromes coronarios, enfermedad vascular cerebral, púrpura trombótica trombocitopénica) en respuesta a concentraciones bajas de agonista. Por ejemplo, en síndrome de insuficiencia coronaria aguda se ha observado la formación de microagregados plaquetarios (< 100 plaquetas).109
La prueba Verify Now consiste en un agregómetro que monitorea de forma rápida y simple la función de los anti Gp IIb/IIIa y de otros antiagregantes plaquetarios.110 Es de vital importancia censar la función de estos fármacos porque en estudios recientes (EPIC, EPILOG) se sugiere que se necesita un bloqueo de receptores plaquetarios mayor a 80% para obtener eficiencia clínica con el uso de antiagregantes plaquetarios.89
La metodología consiste en sets ricos en fibrinógeno, que se utilizarán para aglutinar en sangre total, en relación con la cantidad de receptores plaquetarios ocupados por el antiagregante a estudiar. Para esta prueba se utilizan cartuchos con fibrinógeno y activadores plaquetarios (péptido activador del receptor de trombina para abciximab, ácido araquidónico para ácido acetilsalicílico y ADP para P2Y12).111
Se coloca sangre total en el cartucho, lo que ocasionará que el agonista active las plaquetas, que se expondrán al antiagregante a estudiar y el fibrinógeno se unirá a las plaquetas no ocupadas por el antiagregante, de esta forma se obtiene como resultado el porcentaje de inhibición plaquetaria en pocos minutos.
Agregometría plaquetaria en la práctica clínica
La utilidad de estos estudios es muy amplia; sin embargo, los resultados no son específicos de algún trastorno en particular y, por el contrario, puede haber sobreposición de entidades con un mismo patrón de agregación plaquetaria.
Por lo anterior, se requiere el conocimiento amplio de todas las variables que intervienen en el desarrollo de la prueba y los resultados posibles. La agregometría plaquetaria no debe utilizarse como estudio general sin una indicación o sospecha específica.
Ante una trombocitopenia menor a 100 x 109/L, la agregometría plaquetaria no dará resultados válidos o confiables y deberá esperarse hasta tener una cifra de plaquetas mayor para realizar la prueba.38 El estudio de los trastornos funcionales de las plaquetas puede dividirse de manera general en trastornos hereditarios o primarios y adquiridos o secundarios.
Los defectos primarios de la función plaquetaria son poco frecuentes; por el contrario, los trastornos adquiridos son muy comunes y se asocian con enfermedades cotidianas.38
A continuación se describen los padecimientos más frecuentes y las gráficas de la agregometría plaquetaria correspondientes.
Disfunción plaquetaria primaria (hereditaria)
Síndrome de Bernard-Soulier. Trastorno autosómico dominante, en el que las plaquetas carecen de las glicoproteínas de membrana GP Ib, V y IX. Al haber carencia de glicoproteínas, las plaquetas son incapaces de unirse con el factor de von Willebrand volviéndose incapaces de llevar a cabo la adhesión plaquetaria. Las manifestaciones clínicas principales son sangrado fácil (al mínimo traumatismo), epistaxis, petequias, púrpura e hiperpolimenorrea.39-41
Los hallazgos de laboratorio muestran un patrón de adhesión alterado y agregación anormal con ristocetina (Figura 7); el frotis de sangre periférica contiene plaquetas gigantes y trombocitopenia leve en la gran mayoría de los pacientes.41,42 El diagnóstico diferencial es con enfermedad de von Willebrand. La diferencia entre estas dos enfermedades se evidencia al estudiar la actividad de coagulación del factor VIII (VIII:c), el factor VIII antigénico y la actividad del cofactor VIII: factor de von Willebrand.43

Figura 7 Agregometría de un paciente con síndrome de Bernard-Soulier. La curva de la agregación con ristocetina también es característica de la enfermedad de von Willebrand.
Enfermedad de von Willebrand (EvW). La enfermedad de von Willebrand es la coagulopatía hereditaria más común en todo el mundo, con incidencia de 1 a 4%, sin aparente predominio étnico o racial.45,46 Se trata de un trastorno variable del factor de von Willebrand, ya sea cuantitativo o cualitativo descrito en 1926 en Suecia por Erik von Willebrand.44 Esta enfermedad es el trastorno hemorrágico primario de mayor frecuencia e importancia clínica. Su diagnóstico no siempre es sencillo y requiere tres criterios: antecedentes de hemorragia, disminución en la cantidad o alteración funcional del factor de von Willebrand y demostrar su patrón hereditario.47
El factor de von Willebrand se produce en las células endoteliales y en los megacariocitos; actúa mediante la unión a la colágena subendotelial a través de la adhesión con la GP Ib y GP IIb/IIIa de las plaquetas. También participa de manera decisiva en la coagulación, al proteger al factor VIII:c de la degradación proteolítica de la proteína C, por lo que aumenta la vida media del factor VIII circulante.48
La manifestación clínica es muy variable y es consistente entre familias. El dato clínico característico es la hemorragia “plaquetaria” (sangrado al mínimo traumatismo, hemorragia mucocutánea, menorragia). El espectro de la enfermedad va de leve a severo. Los pacientes con enfermedad leve usualmente son asintomáticos, excepto en situaciones de reto hemostático (traumatismo o cirugía, incluso con procedimientos menores). Los casos severos pueden parecer clínicamente idénticos a la hemofilia.
La enfermedad de von Willebrand puede dividirse en tres tipos principales. El tipo 1 es una deficiencia cuantitativa de los multímeros de todos los tamaños del factor de von Willebrand; el tipo 2 consiste en déficit cualitativo y cuantitativo de los multímeros de alto peso molecular del factor de von Willebrand (en la mayor parte de los subtipos) y el tipo 3 se caracteriza por la ausencia casi completa del factor de von Willebrand.49
El estudio de un paciente en quien se sospecha enfermedad de von Willebrand requiere realizar pruebas in vivo e in vitro. El tiempo de sangrado se utiliza para valorar la hemostasia primaria, pero este estudio in vivo carece de sensibilidad en casos leves y tiene escasa reproducibilidad. La determinación de la actividad del cofactor del factor de von Willebrand ristocetina mide la actividad funcional del factor de von Willebrand mediante el uso de ristocetina para inducir un cambio conformacional en el factor de von Willebrand presente en el plasma del paciente que agrega las plaquetas mediante la unión a su receptor, la GP Ib (Figura 7). También se requiere la cuantificación del factor de von Willebrand antigénico mediante métodos inmunológicos y el análisis de los multímeros de factor de von Willebrand por inmunoelectroforesis en gel de agarosa.
Trombastenia de Glanzmann y atrombia esencial. En estos trastornos las plaquetas carecen de la GP IIb/IIIa y son incapaces de efectuar la unión entre GP IIb/IIIa y fibrinógeno, lo que impide por completo la agregación plaquetaria.
El cuadro clínico será similar al de cualquier otro paciente con disfunción plaquetaria, con la peculiaridad que la tendencia hemorrágica disminuye en severidad con la edad. Las alteraciones bioquímicas incluyen un tiempo de sangrado prolongado, ausencia total de la agregación primaria inducida por ADP, trombina, colágena y epinefrina (Figura 8), deficiencia de factor 3 plaquetario y retracción del coágulo anormal.38,51,52 La única diferencia con la atrombia esencial es que en esta enfermedad la retracción del coágulo es normal.53

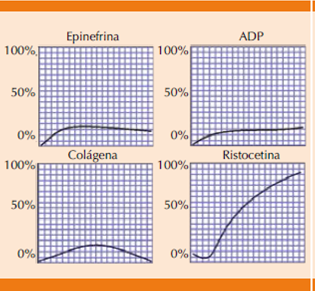
Enfermedad de la poza de almacenamiento. Los trastornos secundarios de la agregación son, por mucho, más comunes que los primarios, de los secundarios el más común es la enfermedad de la poza de almacenamiento. Este trastorno puede formar parte de otros síndromes congénitos, como el de Wiskott-Aldrich, de Chediak-Higashi, de Hermansky-Pudlak y el de trombocitopenia y ausencia de radio (síndrome TAR).
Esta enfermedad se caracteriza por deficiencia de los gránulos densos, que pueden detectarse por microscopia electrónica o por medio de la agregometría plaquetaria, en la que hay ausencia de la curva de agregación secundaria con ADP y epinefrina, y con colágena la curva es marcadamente aplanada o está ausente (Figura 9). El cuadro clínico es inespecífico y la hemorragia es variable, pero severa cuando ocurre.54,55
Disfunción plaquetaria adquirida
Estos trastornos se distinguen por disfunción plaquetaria adquirida, representan la principal causa de hemorragia. Por lo general, estos pacientes tienen sangrados profusos porque no son diagnosticados y se someten a cirugías o sufren traumatismos.
En el Cuadro 3 se recapitulan las principales causas de disfunción plaquetaria adquirida.
Cuadro 3 Principales causas de disfunción plaquetaria adquirida
|
Medicamentos Uremia Enfermedad hepática Paraproteinemias Enfermedades autoinmunitarias Productos de degradación de la fibrina (y fibrinógeno) Síndromes mieloproliferativos/síndromes mielodisplásicos Cirugía cardiaca con circulación extracorpórea |
Disfunción plaquetaria en uremia. La mayoría de los pacientes urémicos tendrán disfunción plaquetaria56 secundaria al aumento en la circulación de los ácidos guanidinosuccínico o hidroxifenólico que interfieren con la actividad del factor plaquetario 3.57,58 Ambos ácidos son dializables corrigiendo o mejorando la función plaquetaria. También puede encontrarse anormalidad en la interacción entre el factor de von Willebrand y los receptores de membrana, principalmente con la GP IIb/IIIa, lo que sugiere un defecto funcional en la interacción adhesiva.59,60
En cuanto a la agregometría plaquetaria, los patrones no son uniformes o característicos y se puede encontrar cualquier combinación de defectos (Figura 10 ).

Disfunción plaquetaria en insuficiencia hepática. La insuficiencia hepática aguda y crónica muestra de manera frecuente disfunción plaquetaria, generalmente secundaria a causas multifactoriales. En los pacientes con enfermedad hepática crónica la activación primaria del sistema fibrinolítico (fibrinólisis primaria) resulta en aumento de los productos de degradación de la fibrina (PDFs), mismos que afectan gravemente la función de las plaquetas, por bloqueo de los receptores de superficie y por disminución de la unión de fibrinógeno con la GP IIb/IIIa.61,62
Otra causa de disfunción plaquetaria en enfermedad hepática es el aumento en el número de plaquetas viejas circulantes, que se observa con la disminución del volumen plaquetario medio (VPM); se cree que estas plaquetas tienen menor actividad hemostática.63 En estos pacientes existen defectos de la agregación secundaria o alteraciones tipo enfermedad de la poza de almacenamiento (Figura 9) que se manifiestan por curvas de agregación aplanadas para colágena, trombina y ristocetina y ausencia de las curvas de agregación secundaria con ADP y epinefrina.61
La afección plaquetaria será mayor si el paciente abusa del alcohol. La ingestión de dosis altas de alcohol, aun sin enfermedad hepática, puede inducir un defecto tipo enfermedad de la poza de almacenamiento, con disminución de ATP y ADP por efecto directo del alcohol y de AMP por inhibición de la adenil ciclasa. También se altera la síntesis de prostaglandinas, TXA2 y la disponibilidad del factor 3 plaquetario.64
Disfunción plaquetaria en paraproteinemias. Muchos pacientes con mieloma múltiple, macroglobulinemia de Waldenström o gammapatías monoclonales tienen de manera concomitante un trastorno de la función plaquetaria. Esto se debe a que las paraproteínas envuelven o cubren a las plaquetas e impiden el contacto de los receptores de membrana con sus ligandos. Esta disfunción plaquetaria también puede suceder con hiperproteinemias de origen benigno.65,66
La mayoría de los pacientes con paraproteinemias malignas manifiestan disfunción plaquetaria significativa con sangrado y alteración difusa de la agregometría plaquetaria (Figura 11). Estas alteraciones son reversibles de manera parcial con plasmaféresis o posterior al control de la actividad de las células plasmáticas con tratamiento específico.67,68

Disfunción plaquetaria en mielodisplasia y síndromes mieloproliferativos. La función plaquetaria está alterada de manera frecuente en los síndromes mieloproliferativos (trombocitemia esencial, metaplasia mieloide agnogénica, policitemia vera, leucemia mieloide crónica) y en los mielodisplásicos (anemia resistente, anemia resistente con exceso de blastos y anemia sideroblástica).69,70 Los patrones de agregación plaquetaria que se asocian con estos síndromes por lo general no son característicos y puede ocurrir casi cualquier combinación de curvas en la agregometría plaquetaria. Los más frecuentes son los defectos de la agregación con epinefrina, seguida de la hipoagregabilidad para ADP.69-71
Disfunción plaquetaria en cirugía cardiaca. De todos los trastornos adquiridos la disfunción plaquetaria secundaria al uso de circulación extracorpórea es por mucho la más severa, es la principal causa de hemorragia trans y posoperatoria en cirugía cardiaca.72,73
Son muchos los factores que se asocian con estas alteraciones plaquetarias: pH, cifra de plaquetas, hematócrito, fármacos, productos de degradación de la fibrina, manejo de la bomba de circulación extracorpórea (velocidad, tiempo, anticoagulación, calcio), oxigenación, hipotermia.72-74 De igual forma, se ha demostrado desgranulación selectiva de plaquetas en cirugía cardiaca; sin embargo, no se han establecido con precisión todos los mecanismos que pueden estar implicados. Sea cual sea el o los mecanismos responsables, el grado de disfunción plaquetaria en circulación extracorpórea es severo y se manifiesta en todos los pacientes.74,75 De igual forma es frecuente que los pacientes a quienes se les realicen estas cirugías ingieran fármacos antiagregantes plaquetarios (por ejemplo: aspirina, clopidogrel, dipiridamol, AINES) que interfieren con la función plaquetaria y que aumentan el riesgo de hemorragia trans y posoperatoria en comparación con los pacientes que no ingieren estos fármacos, por lo menos durante los 10 días previos a la intervención quirúrgica.
Disfunción plaquetaria inducida por fármacos. Existe una gran cantidad de fármacos que causan disfunción plaquetaria, cuya disfunción puede ser evidenciada por medio de la agregometría plaquetaria. Los tres mecanismos más importantes por medio de los que los fármacos alteran el funcionamiento de las plaquetas, en orden de frecuencia son: a)interferencia del fármaco con la membrana o receptores de las plaquetas, b)inhibición de la biosíntesis de prostaglandinas, y c)interferencia con la actividad de la fosfodiesterasa.76,77 Los antiinflamatorios no esteroides (AINES) son los fármacos con actividad antiplaquetaria de administración más frecuente, entre ellos destacan por su mayor consumo: aspirina, ibuprofeno, indometacina, sulfinpirazona, colchicina, dipirona y metamizol. Cualquier AINE puede inducir disfunción plaquetaria importante, y todos actúan mediante la inhibición de las vías de la síntesis de prostaglandinas.78 La aspirina se prescribe para la supresión farmacológica de la función plaquetaria mediante la inhibición de la ciclooxigenasa y, en consecuencia, se inhibe la síntesis de TXA2. Este fármaco también inhibe la síntesis endotelial de prostaciclina. La selectividad inhibitoria es de aproximadamente 70% para la ciclooxigenasa de plaquetas y 30% para la ciclooxigenasa del endotelio, predomina el efecto antitrombótico.79-82
Algunos fármacos psiquiátricos inducen disfunción plaquetaria, como las fenotiazinas (clorpromazina, trifluoperazina, antidepresivos tricíclicos y los inhibidores de la recaptura de serotonina). Varios fármacos que se prescriben en la medicina cardiovascular interfieren con la función de las plaquetas y son causa frecuente de hemorragia anormal (por ejemplo, clofibrato, dipiridamol, ácido nicotínico, papaverina, propranolol, antagonistas de los canales del calcio -nifedipina, verapamil, diltiazem, amlodipino-, bloqueadores β-adrenérgicos, nitroprusiato de sodio y trimetorfan).83-85 Es importante conocer la disfunción plaquetaria causada por estos fármacos porque aumenta de forma importante el riesgo de sangrado trans o posoperatorio en pacientes intervenidos quirúrgicamente.86-88
Asimismo, los antibióticos pueden inducir disfunción plaquetaria, los más frecuentes son las penicilinas, principalmente nafcilina, ampicilina, carbenicilina (el más frecuente) y ticarcilina. La gentamicina y otros aminoglucósidos también son capaces de interferir con la función plaquetaria.30 Existen otras drogas y fármacos que causan disfunción plaquetaria y que aumentan el riesgo de sangrado, entre ellos se encuentran los anestésicos locales (incluida la cocaína), los gases anestésicos, antihistamínicos, dextranes, furosemida, guayacolato de glicerol (base para la mayor parte de los jarabes antitusivos), nitroprusiato, vincristina y vinblastina.30
Los fármacos prescritos para el tratamiento de los síndromes coronarios agudos y durante todas las fases de angioplastia coronaria deben ser estudiados con mayor atención debido a su administración frecuente, a su mecanismo de acción y a su riesgo de causar hemorragia.
Inhibidores del receptor GP IIb/IIIa. Son antiagregantes plaquetarios intravenosos y actualmente se cuenta con tres en el mercado: abciximab, tirofiban y eptifibatide. Abciximab es un anticuerpo monoclonal antirreceptor IIb/IIIa, tirofiban es un inhibidor no peptídico y eptifibatide es un heptapéptido cíclico no inmunogénico que se deriva de veneno de serpiente.89-92
El riesgo de hemorragia es inherente al mecanismo de acción; sin embargo, este riesgo es mayor (dos a tres veces) cuando se combinan con aspirina, clopidogrel, heparina y anticoagulantes orales (por lo general, se combinan dos, tres e incluso cuatro de estos fármacos en un mismo paciente).93-95 La mayor parte de los episodios de sangrado sobrevienen en las angioplastias coronarias en el sitio del acceso vascular, aunque son frecuentes las hemorragias de tubo digestivo y en el sistema nervioso central. El bloqueo del receptor IIb/IIIa induce un estado trombasténico con afección principalmente de la agregación plaquetaria mediada por fibrinógeno. También ocurre trombocitopenia, en ocasiones grave, lo que aumenta la morbilidad de manera significativa y el riesgo de hemorragias fatales.96,97
Tienopiridinas: las tienopiridinas clopidogrel y ticlopidina son fármacos con amplia prescripción en enfermedad cardiovascular que antagonizan de manera selectiva y diferente a la aspirina y AINES el efecto de ADP. Ambos fármacos inhiben la agregación plaquetaria ex vivo y prolongan el tiempo de sangrado in vivo. El grado de prolongación del tiempo de sangrado es equivalente o mayor al que produce la aspirina y el efecto de la combinación de tienopiridina y aspirina es aditivo. Las curvas de agregometría muestran una clara inhibición para ADP y epinefrina, mientras que las respectivas para trombina y colágena son normales.98,99
Hiperfunción plaquetaria. Los estados de hiperactividad plaquetaria hereditarios o adquiridos son causa de un número importante de eventos trombóticos arteriales y algunos venosos. Un ejemplo es el defecto Wein-Penzing; éste es poco frecuente y fue descrito en 1991. Se trata de una deficiencia en la vía de la lipooxigenasa con incremento compensador de los productos de la ciclooxigenasa (TXA2, PGE2 y PGD2) que se asocia con infartos de miocardio en gente joven.100 Otro defecto con hiperfunción plaquetaria, muy común y de fácil diagnóstico y tratamiento, es el síndrome de la plaqueta pegajosa, causante de gran número de episodios de trombosis arterial y venosa (hasta 14% de los casos de trombofilia primaria) con morbilidad y mortalidad significativas.101,102 Se hereda con carácter autosómico dominante103 y las principales manifestaciones clínicas son trombosis coronaria y vascular cerebral en personas jóvenes sin otros factores de riesgo cardiovascular, aunque puede haber episodios trombóticos arteriales y venosos en cualquier sitio.104,105 Los criterios de diagnóstico descritos por Mammen103 se señalan en el Cuadro 4. El diagnóstico se establece mediante la agregometría plaquetaria en plasma rico en plaquetas con la adición de ADP y epinefrina a diferentes concentraciones (Cuadro 5). Se considera diagnóstico de hiperactividad plaquetaria y síndrome de la plaqueta pegajosa con:
Cuadro 4 Síndrome de la plaqueta pegajosa: criterios diagnósticos
|
Síndrome de la plaqueta pegajosa Tipo I: hiperagregabilidad a epinefrina y adenosín difosfato. Tipo II: hiperagregabilidad a epinefrina únicamente Tipo III: hiperagregabilidad a adenosín difosfato únicamente Diagnóstico sospechoso: Hiperagregabilidad a sólo un agonista a una concentración y antecedentes de trombosis |
Cuadro 5 Concentraciones de los agonistas y valores normales de la agregometría
| Agonista | Variación normal (porcentaje de agregación) |
|---|---|
| EPI: 11 x 10-6 M | 39-80 |
| EPI: 1.1 x 10-6 M | 15-27 |
| EPI: 0.55 x 10-6 M | 9-20, 7.5-55, 2-36, 0-12 |
| ADP: 2.34 x 10-6 M | 7.5-55 |
| ADP: 1.17 x 10-6 M | 2-36 |
| ADP: 0.58 x 10-6 M | 0-12 |
EPI: epinefrina; ADP: adenosín difosfato.
1)Antecedente de trombosis e hiperagregabilidad plaquetaria a dos concentraciones de un agonista, o 2)antecedente de trombosis e hiperagregabilidad plaquetaria a una concentración de dos agonistas, o 3)antecedente de trombosis e hiperagregabilidad plaquetaria a una concentración de un agonista y al repetir la prueba se obtienen los mismos resultados.
El tratamiento consiste en la administración de antiagregantes plaquetarios, principalmente aspirina, y es de suma importancia verificar el efecto farmacológico (antiagregante) en la agregometría plaquetaria.
En conclusión, la agregometría plaquetaria y sus derivados son el patrón de referencia para el diagnóstico de múltiples trastornos funcionales plaquetarios primarios o adquiridos y su conocimiento es fundamental en el actuar del médico internista.

