











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkANTECEDENTES
La palabra alcohol comúnmente se usa para referirse específicamente al etanol o alcohol etílico. Sin embargo, en térmicos químicos, el alcohol puede referirse a cualquier derivado de un hidrocarburo. Por extensión, el sufijo "ol" se usa en la nomenclatura química para designar cualquiera de varios alcoholes. Los hidrocarburos que tienen dos grupos hidroxilos son llamados glicoles. El glicol derivado del etano es llamado etilenglicol.1 La producción estimada en todo el mundo excede 23 mil millones de kg anualmente. El etilenglicol, 1-2 etanediol tiene un peso molecular de 67.2 g/mol. Es un líquido viscoso, no volátil, incoloro, higroscópico, de sabor dulce, soluble en agua.2,3 Se usa como el principal ingrediente en muchos productos anticongelantes automotrices, esas formula ciones pueden contener varios aditivos, como anticorrosivos, lubricantes, agentes antiespuma y tintes. La fluoresceína sódica, un compuesto que exhibe fluorescencia cuando se expone a la luz ultravioleta, es común, pero no universalmente adicionado para ayudar a identificar fugas en los sistemas de enfriamiento de automóviles. El benzoato de denatonio, un derivado de la lidocaína, con un intenso sabor desagradable, se adiciona a algunos anticongelantes para disuadir la ingesta accidental o intencional. El uso industrial más importante es como precursor sintético. Por ejemplo, el taraftalato de polietileno, el polímero usado para hacer botellas de plástico para las bebidas carbonatadas y otros contenedores, es producido del etilenglicol, así como el textil sintético familiar conocido como poliéster. El etilenglicol por sí solo puede resultar en envenenamientos que ponen en peligro la vida cuando es ingerido. La utilidad del etilenglicol deriva de su baja masa molecular, su mezcla con el agua y su alto punto de ebullición.1 El etilenglicol empezó a usarse ampliamente en el decenio de 1920; el primer caso por envenenamiento se describió en 1930. La toxicidad de los glicoles se apreció completamente en 1937 cuando 76 personas murieron después del uso de un elixir de sulfanilamida que contenía 72% de etilenglicol. En 1969 siete niños murieron en Ciudad del Cabo, Sudáfrica, como resultado de la ingesta de un hipnótico que también contenía esta sustancia. La Asociación Americana de los Centros de Control de Envenenamientos reportó más de 4,800 y 6,000 exposiciones a etilenglicol en 1997 y 1998, respectivamente.3,4
CASO CLÍNICO
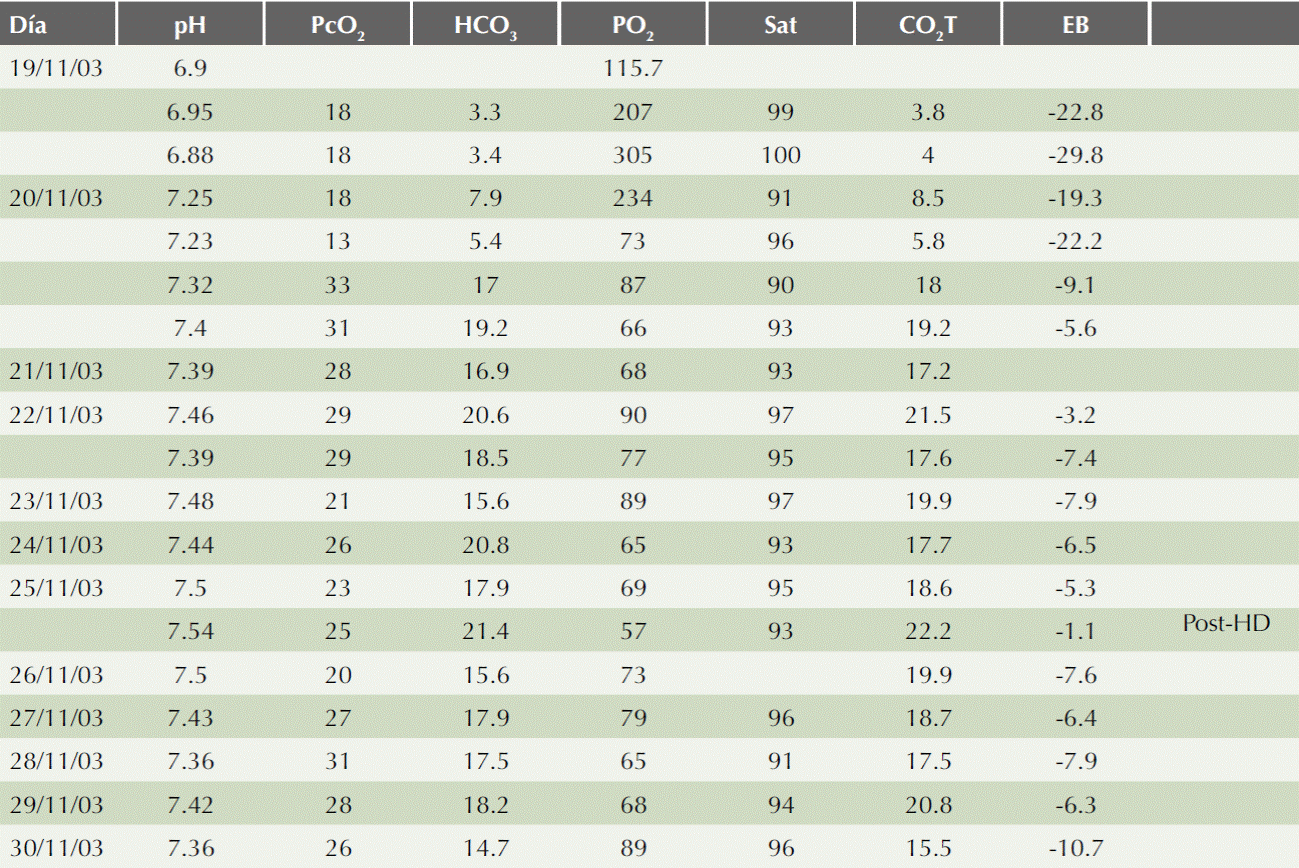
Paciente masculino de 90 años de edad, originario y residente del estado de Tlaxcala, jornalero. Con alcoholismo positivo desde los 25 años de edad a base de bebidas fermentadas, llegando frecuentemente a la embriaguez. En 1998 cursó con enfermedad vascular cerebral oclusiva de la arteria cerebral media izquierda que le dejó como secuela hemiparesia espástica derecha. Inició su cuadro clínico en noviembre de 2003, había ingerido presumiblemente 500 cc de líquido anticongelante que se encontraba en una botella de refresco, con verborrea, poliuria y polidipsia. Un día después se agregó somno lencia, que evolucionó al deterioro neurológico, así como respiración estertorosa. Fue llevado 24 horas después de la ingesta a su Hospital General de Zona correspondiente. Dos horas antes de su llegada a su hospital de adscripción tuvo pérdida del estado de alerta, además de respiración rápida y superficial; a su llegada se encontró con pérdida del estado de alerta, respiración acidótica y ruidos cardiacos arrítmicos. La gasometría tomada a su ingreso reportó pH de 6.9 sin determinación de PCO2 ni de bicarbonato. Por deterioro de la mecánica ventilatoria se decidió intubación orotraqueal y asistencia mecánica ventilatoria. Fue trasladado a la Unidad de Cuidados Intensivos de nuestra unidad, donde se encontró bajo asistencia mecánica ventilatoria; se tomó control gasométrico que mostró acidosis metabólica con brecha aniónica de 35.79; la citometría hemática reportó leucocitosis con neutrofilia, mientras que la química sanguínea reportó hiperglucemia, azoemia e hiperfibrinogenemia. A pesar de reposición de bicarbonato, persistió con acidosis metabólica por lo que se le realizó diálisis peritoneal. Posteriormente padeció crisis convulsivas tónico-clónico generalizadas, anuria y agitación psicomotriz. Continuó con acidosis metabólica a pesar de diálisis peritoneal y reposición de bicarbonato. El Cuadro 1 muestra los resultados de laboratorio y el Cuadro 2, los resultados de las gasometrías. Se solicitó apoyo al centro toxicológico de Monterrey; a pesar del tiempo transcurrido, se decidió la administración de etanol por sonda nasogástrica a razón de 0.5 mL/kg/dosis, cada dosis se administró cada cuatro horas. Veinticuatro horas después de haberse iniciado la administración de etanol, el paciente se encontró soporoso y con localización del estímulo nociceptivo. Se sometió a hemodiálisis al sexto y séptimo días de haber ingresado por mayor incremento en las concentraciones de los elementos azoados. Después de la segunda sesión de hemodiálisis se encontró despierto, colaborador y obedeciendo órdenes. Se retiró el soporte ventilatorio a los 11 días; se egresó a los 13 días con persistencia de la elevación de los azoados.

DISCUSIÓN
El etilenglicol (1,2-etanediol, etano-1,2-diol) es un alcohol con dos grupos hidroxilo. La fórmula molecular es C2H6O2, su masa molar es de 62.7 g/mol, densidad 1.132 g/cm3. En su forma pura, es un líquido sipuroso, incoloro, inodoro, con sabor dulce y viscosidad de 16.1 mPas. Es un líquido relativamente no volátil, densidad de vapor 2.14 (aire=1), completamente soluble en agua en todas proporciones. Soluble en alcoholes alifáticos bajos, acetona, glicerol y ácido acético, algo soluble en éter, benceno y aceites. Es un líquido inflamable con punto de combus tión de 127°C y temperatura de autoignición de 398°C, limites explosivos de 3.2-53%. Es manufacturado a partir del etileno por vía intermedia de óxido de etileno, que reacciona con agua para formar etilenglicol.5
Toxicinética
La exposición dérmica en actividades como cambiar anticongelantes es la vía de exposición más común del etilenglicol; sin embargo, es más factible que ésta no lleve a efectos tóxicos, ya que la absorción significativa de etilenglicol a través de la piel intacta no ocurre con la exposición casual. La exposición por inhalación se debe a su baja volatilidad; sin embargo, la presión de vapor del etilenglicol es también baja como para llevar a la exposición excesiva por inhalación a temperaturas incrementadas o rociadas en forma de aerosol. Empero, las concentraciones aéreas altas de etilenglicol teóricamente pueden resultar en absorción por inhalación significativa que rápidamente causa irritación intolerable de la garganta y de las membranas mucosas y, por ende, previene la exposición significativa.1,5 Sólo la exposición oral, a través de la ingesta intencional o accidental, tiene efectos tóxicos y sólo si es ingerido en cantidades suficientes en una sola exposición. Es absorbido rápida y com pletamente después de su ingesta por el aparato gastrointestinal y alcanza concentraciones pico entre 30 y 60 minutos después de su ingesta,5 con concentraciones máximas que se alcanzan de una a cuatro horas.2,6 La vida media es de 2.5-4.5 horas, puede prolongarse hasta 17 horas en presencia de concentraciones terapéuticas de etanol (100-200 mg/dL).5 Tiene baja afinidad por proteínas plasmáticas debido a su hidrosolubilidad, lo que favorece una tasa alta de distribución tisular. Su volumen de distribución es de 0.54 0.8 L/kg, similar al agua corporal total.1 2 5 6 Su metabolismo es principalmente hepático (80%) con vida media corta de tres a ocho horas.12 Es filtrado por el glomérulo renal y reabsorbido pasivamente; cerca de 20% es filtrado por los ríñones, pero la tasa de excreción a través de esta vía es lenta, con vida media de 18-20 horas.5
La dosis mínima letal no está bien establecida. La información disponible sugiere que hay una considerable variación persona a persona. Hay varios factores conocidos que pueden afectar esta variabilidad, que incluyen el grado de dilución del etilenglicol contenido en la formulación específica ingerida, la coingesta, principalmente de etanol, porque puede influir en la respuesta a un volumen dado de ingestión; si ocurre vómito y la función renal.1 La dosis tóxica letal es citada comúnmente como de 100 mL o de 1-1.5 mL/ kg, pero también tiene una variabilidad considerable.1, 2,4,7 Cuando se trata apropiadamente, los pacientes pueden sobrevivir a pesar de ingestas mayores.4 Estos reportes se basan en experi mentos animales o en reportes clínicos, algunas veces anecdóticos y probablemente implican varios grados de desconocimiento o diluciones no especificadas, más que de etilenglicol puro. En nuestro caso, la información disponible es que el paciente ingirió 500 mL, lo que es poco probable ya que hubiese fallecido al exceder la dosis letal o, como se señala, el etilenglicol probablemente estaba diluido.
Similar al etanol, tiene efectos de ebriedad y sedación del sistema nervioso central, pero la toxicidad inherente de este compuesto perse es baja. Sin embargo, el compuesto es oxidado a metabolitos intermediarios que poseen citotoxicidad sustancial. Se metaboliza principalmente por enzimas deshidrogenasa, en conjunto con la forma oxidada del cofactor nicotinamida adenina dinucleótido (NAD+). La primera es catabolizada por la alcohol deshidrogenasa, que es capaz de oxidar ciertos átomos que contienen hidroxilos para aldehidos. Una segunda enzima, la aldehido deshidrogenasa, convierte el aldehído a su correspondiente ácido carboxílico.

Esta reacción constituye uno de dos importantes pasos limitantes de la vía metabòlica total. El glicoaldehído rápidamente se somete por oxidación a aldehido en parte por la aldehido deshidrogenasa y en menor extensión por la enzima citocromo P450 (CYP2E1). El aldehido inhibe la fosforilación oxidativa, la respiración, el metabolismo de la glucosa, la síntesis de proteínas, la replicación de ADN, la síntesis de ARN ribosomal, la respiración en el sistema nervioso central, el metabolismo de la serotonina, además de alterar las concentraciones de aminas en el sistema nervioso central, resultando en la producción de glioxal y de ácido glicólico.
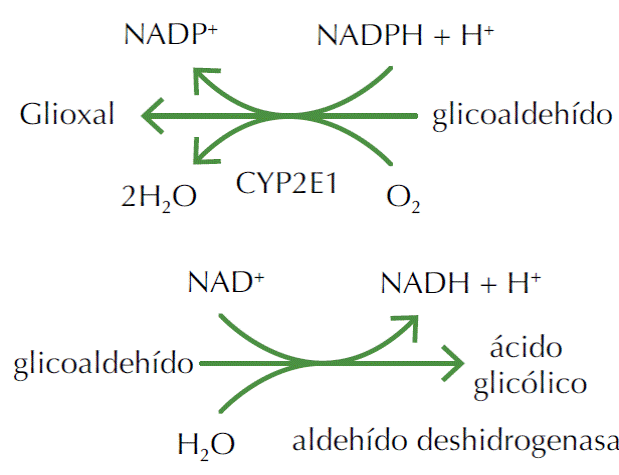
El glioxal es metabolizado a ácido glicólico y a ácido glioxílico. El ácido glicólico es el responsable de la acidosis metabòlica.

Enseguida, el ácido glicólico es convertido a ácido glioxílico por la glicolato oxidasa o lactato deshidrogenasa, una conversión que constituye el segundo paso limitante de la secuencia total. El ácido glioxílico es convertido a ácido oxálico principalmente por la DHL, pero también en algún grado por la glicolato oxidasa.

Los ácidos glicólico, glioxílico y oxálico se disocian principalmente bajo condiciones fisiológicas, liberando iones hidrógeno de su ácido carboxílico. Los modelos animales y autopsias de humanos han revelado formación extensa de cristales visibles por microscopia de luz en varios tejidos, que incluyen túbulos renales, cerebro, meninges, vasos sanguíneos, hígado, bazo, pericardio y el sistema de conducción cardiaca. Estos cristales se han identificado como de oxalato de calcio, la sal de calcio escasamente soluble de ácido oxálico, que se forma en soluciones acuosas de ácido oxálico en presencia de iones de calcio.

El oxalato de calcio no puede ser metabolizado por los humanos y es un producto final del metabolismo del etilenglicol.1,3,4,7-9
El hallazgo patológico de grandes formaciones de oxalato de calcio en los túbulos renales fue aceptado grandemente como el mecanismo por el cual el envenenamiento por etilenglicol resulta en las manifestaciones clínicas de insuficiencia renal aguda, ya sea por causar obstrucción tubular renal o por un efecto citotóxico directo del oxalato en los túbulos renales. Este mecanismo tiene una presunción lógica, dado que los individuos con el error innato del metabolismo conocido como oxaluria primaria u oxalosis congènita excretan grandes cantidades de oxalato de calcio y desarrollan un extenso depósito de cristales, incluyendo nefrocalcinosis, lo que lleva a insuficiencia renal frecuentemente en niños.
Además de la nefropatia microobstructiva por el oxalato de calcio o la necrosis tubular aguda inducida por oxalato, otros mecanismos postulados de toxicidad renal por el envenenamiento con etilenglicol han incluido la citotoxicidad renal directa por uno o más de los metabolitos intermediarios, necrosis focal hemorrágica cortical, otros mecanismos de necrosis tubular aguda y nefritis intersticial. El glicoaldehído y el glioxilato mostraron alto grado de toxicidad, incluyendo depleción profunda de ATP, liberación de DHL y muerte celular tubular. El glicoaldehído causa también degradación de fosfolípidos y de la DHL. El glioxilato mostró inhibir el transporte mitocondrial de electrones y la fosforilación oxidativa, inhibiendo el ciclo de Krebs. El glioxal también tiene citotoxicidad con formación de especies reactivas de oxígeno y colapso del potencial de membrana mitocondrial, incluyendo peroxidación lipídica e inhibiendo la respiración celular. Los intermediarios ácidos y aldehídos de etilenglicol tienen otros efectos tóxicos, como interferir en el metabolismo de la glucosa, bloquean la síntesis de proteínas e inhiben la síntesis y replicación del ácido nucleico.1
Una hipótesis reciente de trabajo es que el oxalato, el metabolito del etilenglicol, se acumula en el túbulo renal proximal en cantidades suficientes para formar cristales de monohidrato de oxalato, que son introducidos por las células proximales tubulares, lo que provoca daño mitocondrial que resulta en muerte celular y necrosis tubular con la consecuente insuficiencia renal. Pomara y colaboradores demostraron en ratas que los cristales de monohidrato de oxalato de calcio y no el ion oxalato causan daño en la mitocondria del riñón, induciendo permeabilidad mitocondrial transitoria que lleva a la muerte de las células renales. Debido a que esos cristales de monohidrato de oxalato de calcio son transportados intracelularmente por las células renales, la toxicidad renal del etilenglicol puede resultar en la inhibición de la respiración mitocondrial en las células tubulares proximales. Se sabe que la permeabilidad transitoria de la mitocondria inhibe la síntesis de ATP y es responsable de la muerte celular necrótica. Así que la interferen cia con la producción de energía mitocondrial puede ser un factor importante en la lesión renal inducida por la acumulación de cristales de monohidrato de oxalato de calcio en los riñones durante la exposición aguda y subaguda al etilenglicol.10
En cerca de 50% de los casos fatales de intoxicación por etilenglicol, se encuentran cristales de oxalato de calcio en la orina y en diferentes tejidos. Kim y colaboradores11 observaron esos cristales en la orina cuatro a ocho horas después de la ingestión. La existencia de cristales de oxalato de calcio depende del curso de la intoxicación. Así, Garg y colegas reportaron el caso fatal de un paciente que tenía concentraciones séricas de etilenglicol de 2,340 mg/dL; la elevada concentración no se debió a la redistribución postmortem, ya que una concentración de 2,261 mg/dL se halló en la orina y en el humor vítreo fue de 1,028 mg/dL; a pesar de estas concentraciones, no se encontraron metabolitos tóxicos ni cristales de oxalato de calcio; además, las concentraciones de calcio, de creatinina y de nitrógeno ureico fueron normales, se asumió que fue consecuencia del efecto tóxico directo del etilenglicol que provocó depresión aguda del sistema nervioso central, insuficiencia respiratoria y muerte antes de la acumulación de metabolitos tóxicos.12
Historia clínica y manifestaciones
El etilenglicol es un tóxico y su ingestión debe considerarse una urgencia médica. El etilenglicol puede ser ingerido de manera accidental o puede ser tomado deliberadamente en un intento suicida o como sustituto del etanol. La intoxicación con esta sustancia requiere tratamiento con un antídoto.5
En 1950, Khan y Brotchner describieron los siguientes estadios clásicos de la intoxicación por etilenglicol: 1) neurológico, 2) cardiopulmonar y 3) renal, algunos autores incluyen un estadio 4 o de secuelas. Estos estadios son fases teóricas de la intoxicación por etilenglicol, pero la progresión del curso clínico con frecuencia es no consistente o predecible. Un estadio puede predominar, mientras que otro puede estar ausente, o bien existir sobreposición de los tres estadios. Los pacientes pueden presentarse muchas horas después de la ingesta y encontrarse comatosos con dificultad respiratoria e insuficiencia renal.6
El primer síntoma de la ingesta de etilenglicol es similar al causado por la ingesta de alcohol, esta ebriedad puede ser indistinguible en el examen físico de la causada por la intoxica ción por etanol. En pocas horas, más efectos tóxicos aparecen, los síntomas pueden incluir náuseas, vómitos, convulsiones, estupor o incluso coma. La toxicidad por etilenglicol debe sospecharse en cualquier paciente que parezca severamente enfermo después de tomar una sustancia desconocida, especialmente si parece ebrio y no tiene olor a alcohol en su respiración.1,5
Estadio 1 (minutos-12 horas)
Sobreviene frecuentemente de 30 minutos a varias horas después de la ingestión. Predomina la toxicidad sobre el sistema nervioso central, inicia con manifestaciones de ebriedad similar a la que ocurre con la intoxicación por etanol. Puede estar acompañada de cefalea, vértigo, euforia y comportamiento extraño. El dolor abdominal y gastrointestinal pueden ocurrir temprano, ya que el etilenglicol como el etanol puede causar irritación gástrica, que resulta en dolor abdominal y en algunos casos en vómitos. A medida que el metabolismo progresa, estos síntomas son reemplazados por acidosis metabólica y depresión del sistema nervioso central. El primer estadio puede persistir más allá de 12 horas, con las manifestaciones neurológicas que empeoran progresivamente. Estas manifestaciones incluyen depresión más severa del estado de conciencia, que en algunos casos puede progresar al coma, asociado con hipotonía e hiporreflexia. Las convulsiones, incluido el estado epiléptico, son comunes en casos severos, junto con un amplio espectro de anormalidades neurológicas que pueden incluir nistagmo, ataxia, parálisis, oftalmoplejías y sacudidas mioclónicas.1-6 La tomografía axial computada y la resonancia magnética nuclear de cráneo muestran edema cerebral e hipodensidades que afectan la médula, puente, cerebro medio, tálamo, diencéfalo, cerebelo, ganglios basales, lóbulos temporales, materia blanca central y periventricular. La meningitis aséptica y encefalitis se han observado también en el examen postmortem.1 Los síntomas neurológicos se deben a los metabolitos aldehídos.3,5 Puede aparecer aumento de la osmolaridad sérica de manera temprana y encontrarse cristales de oxalato de calcio.
Estadio 2 (12-24 horas)
Ocurre típicamente 12 a 24 horas después de la ingestión, se caracteriza por manifestaciones cardiopulmonares. La hiperpnea es evidente en forma temprana en este estadio, como consecuencia de la compensación respiratoria de la acidosis metabólica. En algunos casos, se requiere intubación orotraqueal para proteger la vía aérea por la depresión severa del sistema nervioso central. En otros casos, puede haber edema pulmonar con dificultad respiratoria y ser la causa de insuficiencia respiratoria franca. Puede ocurrir bradicardia, taquicardia, hipo o hipertensión, algunos pacientes pueden padecer insuficiencia cardiaca. Puede sobrevenir choque circulatorio y en algunos casos no responder a la expansión intravascular, requiriendo la administración de vasopresores. También se han señalado neumonitis y sín drome de dificultad respiratoria. Estos efectos cardiovasculares pueden deberse al depósito de cristales de oxalato de calcio en el corazón, pulmones y vasculatura o por efectos directos de otros intermediarios tóxicos en las células de estos sistemas orgánicos.1-6 Se ha identificado disfunción orgánica múltiple en esta fase.13 La muerte ocurre con más frecuencia en esta etapa.
Estadio 3 (más de 24 horas)
Si el paciente sobrevive a las manifestaciones neurológicas y cardiopulmonares, la insuficiencia renal aguda sobreviene con frecuencia, constituyendo la tercera fase de la intoxicación por etilenglicol, que se manifiesta de manera típica entre 24 y 72 horas después de la ingestión. La insuficiencia renal puede ser oligúrica, anúrica o no oligúrica. Los pacientes conscientes pueden quejarse de dolor de flanco. Puede ocurrir hematuria macro o microscópica.1-5 La formación de cristales requiere una cantidad de tiempo suficiente para que el etilenglicol sea metabolizado a oxalato. La formación de oxalato depleta las concentraciones de calcio sérico y se deposita en la mucosa intestinal, el hígado, el cerebro, el corazón, los pulmones y el riñón. La excreción de los cristales de oxalato de calcio está usualmente pero no siempre presente.4 El incremento en la excreción de oxalato ocurre en la intoxicación por etilenglicol. Debido a la baja solubilidad del oxalato en presencia de calcio, con frecuencia se observan cristales de oxalato de calcio en la orina de pacientes intoxicados y pueden ser depositados en las células tubulares y el líquido luminal. Por largo tiempo se ha asumido que estos cristales son la génesis de la insuficiencia renal aguda asociada con el etilenglicol. Sin embargo, los cristales de oxalato de calcio no se han observado en algunos casos de daño renal. Podelski y colaboradores14 estudiaron la citotoxicidad del etilenglicol, glicoaldehído, glicolato, glioxilato y oxalato en segmentos tubulares proximales de ratón y cultivaron células tubulares proximales humanas. Estos autores concluyeron que sólo los metabolitos aldehído, glicoaldehído y glioxilato son tóxicos y sugirieron que cualquier implicación del oxalato en la insuficiencia renal aguda asociada con el etilenglicol es resultado de la cristaluria de oxalato de calcio intraluminal-formación de cilindros. Por el con trario, se ha observado citotoxicidad en células renales humanas cultivadas, sólo por el oxalato de calcio y no por glicoaldehído, glicolato o glioxilato en concentraciones esperadas en la intoxicación por etilenglicol. McMartin presentó evidencia convincente de que la toxicidad renal asociada con etilenglicol se debe a los cristales de monohidrato de oxalato de calcio y no al glicolato, glicoaldehído o glioxilato.15,16
Aunque los estudios viejos sugieren que el daño renal puede ocurrir en ausencia de cristales, la acumulación de cristales de monohidrato de oxalato de calcio recientemente se ligó con evidencia de necrosis tubular proximal. En términos microscópicos, el daño necrótico se observa solamente en presencia de cristales de monohidrato de oxalato de calcio y metabólicamente el daño es más severo en riñones con elevada acumulación de cristales de monohidrato de oxalato de calcio.
El papel del oxalato en la causa de la insuficiencia renal se ha confirmado in vivo en diversos estudios animales, en uno de ellos ratas Wistar y F-344 se alimentaron con una dieta que contenía etilenglicol durante 16 a 52 semanas. Las ratas Wistar fueron más sensibles al etilenglicol que las F-344, ya que casi todas tuvieron nefropatía severa relacionada con cristales, a dosis de 500 mg/kg/día, mientras que las F-344 tuvieron nefropatía mínima o nula. Además, las dosis altas de etilenglicol, 1,000 mg/kg/día en las ratas insensibles, F-344, produce suficientes concentraciones de oxalato de calcio para que ocurra nefropatía. Estos resultados demuestran que la nefropatía tóxica ocurre sólo cuando altas concentraciones de oxalato se acumulan como cristales de monohidrato de oxalato de calcio en los riñones. De manera experimental, la exposición crónica a etilenglicol produce una baja pero constante liberación de oxalato que puede llevar a nefropatía. En estas exposiciones crónicas, la acidosis no ocurre, lo que indica que la toxicidad renal no es secundaria a la acidosis.16-18
Se ha demostrado correlación estrecha entre la severidad de la nefrotoxicidad y la acumulación total de cristales de monohidrato de oxalato de calcio. Estos cristales se adhieren a las membranas de las células tubulares y subsecuentemente ingresan resultando en muerte celular. McMartin sugirió que posiblemente existen factores clave en la formación de cristales de monohidrato de calcio, como la magnitud de la entrega del oxalato y la depuración por el glomérulo.16
Los cristales de oxalato de calcio también son depositados en otros tejidos y presumiblemente son la causa de la toxicidad cerebral y de otros órganos.1
Los posibles mecanismos por los que los cristales de monohidrato de oxalato de calcio inducen la muerte celular incluyen la pérdida de la estructura de la membrana, generación de mediadores lipídicos, desequilibrio entre oxidantes y antioxidantes y disfunción mitocondrial. Inducen redistribución de la fosfatidilserina al lado exterior de la membrana plasmática e incrementan la liberación de ceramida, indicadores tempranos de muerte celular. Producen hemólisis y alteran la integridad de la unión estrecha en las células renales que se han cultivado o insertado para separar los compartimientos basolaterales y apicales sugiriendo daño relacionado con la membrana. El oxalato también activa la fosfolipasa A2, una enzima que produce la señalización de las moléculas de ácido araquidónico y de lisofosfolípidos, que está implicada en la lesión por toxicidad de las células renales.16
La respuesta citotóxica a los cristales de monohidrato de oxalato de calcio puede resultar de estrés oxidativo y de la acumulación incrementada de especies reactivas de oxígeno, acompañadas por un decremento en el potencial antioxidante. Sin embargo, el daño tisular in vivo asociado con hiperoxaluria no se correlaciona con la peroxidación lipídica, por lo que se cues tiona el papel de los mecanismos oxidativos en la producción del daño necrótico. Empero, el oxalato puede ser tóxico a través de efectos oxidativos en las proteínas o los ácidos nucleicos, más que en los lípidos.
Más probablemente, el incremento en las especies reactivas de oxígeno se origina en la mitocondria a través de la pérdida de los complejos I y III de la cadena transportadora de electrones. Los estudios in vivo, efectuados en mitocondrias de ratas, sugieren que el oxalato puede incrementar las especies reactivas de oxígeno y disminuir las proteínas antioxidantes tiol. Los efectos en las especies reactivas de oxígeno son reproducidos por la administración de ácido araquidónico y lisofosfatidilcolina, lo que sugiere que el oxalato puede generar señales moleculares para incrementar la generación mitocondrial de especies reactivas de oxígeno. Los cristales de monohidrato de oxalato de calcio afectan directamente la mitocondria para incrementar la concentración de superóxido, aunque parcialmente a causa de la disociación de los cristales de monohidrato de oxalato de calcio para liberar oxalato, seguido por la captación de oxalato por el transportador mitocondrial dicarboxilato. Entonces, los cristales de monohidrato de oxalato de calcio inducen muerte celular, por incrementar las especies reactivas de oxígeno, ya sea por la acción directa en la mitocondria en la generación de especies reactivas de oxígeno o indirectamente por la generación de moléculas de liposeñalización.
Los químicos con frecuencia son citotóxicos por dañar la mitocondria, comúnmente por incrementar la permeabilidad de la membrana interior a pequeños solutos, esto es, induciendo la conversión de la permeabilidad mitocondrial, asociada con la pérdida del potencial transmembrana, con la inflamación mitocondrial y con la ruptura de la membrana externa. La inducción de la permeabilidad mitocondrial está ligada con cambios en la fluidez de la membrana interna, por tanto, con cambios conformacionales en las proteínas formadoras de poros. La permeabilidad mitocondrial juega un papel clave en la muerte celular necrótica.
La disfunción mitocondrial, al provocar la depleción del ATP intracelular, favorece la producción de necrosis, ya que el ATP se requiere para la inducción de la apoptosis. Así entonces, los cristales de monohidrato de oxalato de calcio también pueden ser citotóxicos por interferir directamente con la respiración mitocondrial y la producción de ATP. Además, los cristales de monohidrato de oxalato de calcio pueden producir toxicidad por un efecto directo en la función mitocondrial, ya que se sabe que cantidades suficientes de cristales de monohidrato de oxalato de calcio son tomados por las células tubulares proximales e interactúan con la mitocondria o con las membranas mitocondriales. En mitocondrias aisladas de riñones de ratas, los cristales de monohidrato de oxalato de calcio, pero no el oxalato, disminuyen marcadamente la respira ción mitocondrial sin alterar el estadio IV de la respiración o la tasa ADP/oxígeno.16
En 50% o menos de los casos de intoxicación por etilenglicol hay cristaluria de oxalato de calcio a la admisión hospitalaria, por tanto, no es un hallazgo de laboratorio muy sensible. Sin embargo, el hallazgo de cristaluria de oxalato de calcio en conjunto con la brecha osmolar y brecha aniónica elevada indican fuertemente toxicidad probable por etilenglicol. Estos cristales están presentes en dos formas, los cristales de dihidrato de oxalato de calcio en forma de envoltura y los cristales de monohidrato de oxalato de calcio termodinámicamente más estables, en forma de aguja, estos últimos se han identificado erróneamente como cristales de hipurato.
Estadio 4 (secuelas)
Aunque no se incluyen en las descripciones clásicas de la intoxicación por etilenglicol, un cuarto estadio (días a semanas posingestión), que se manifiesta por déficits neurológicos retardados, fue originalmente sugerido por Factor y Lava en 1987,19 en tres casos que mostraron déficit de los nervios craneales. Puede ser que con el mejoramiento en los cuidados médicos, más pacientes con sobredosis severas, que anteriormente hubieran resultado mortales, sobrevivan y entonces padezcan secuelas tardías.20
Por lo general, se manifiestan una a dos semanas después de la ingesta inicial e incluyen neuropatías craneales, que pueden ser parálisis facial bilateral y oftalmoplejía, así como también neuropatías periféricas sensitivomotoras, que pueden ser severas para causar parálisis completa. Los reportes también incluyen edema cerebral, convulsiones, aumento de la presión intracraneal con hidrocefalia comunicante, enfermedad vascular cerebral, parálisis diafragmática, puede haber incluso manifestaciones de disfunción del sistema nervioso autónomo como síndrome taquicardia-bradicardia y retención urinaria con íleo paralítico. La punción lumbar muestra disociación albuminocitológica, aunque puede haber leucorraquia. La tomografía computada puede ser normal, pero las imágenes de resonancia magnética nuclear pueden mostrar crecimiento anormal de los núcleos de los nervios craneales. La neurofisiologia puede mostrar polineuropatía axonal primaria, una afección desmielinizante o polirradiculopatía. Rahman y colegas17 reportan el caso de un paciente intoxicado por etilenglicol que padeció como secuelas disfunción del sistema nervioso autónomo con hipotensión postural y gastroparesia; asimismo, Baldwin y colaboradores reportaron el caso de un paciente que ingirió etilenglicol y padeció parálisis bulbar, hipotensión postural y dolor neuropático persistente.21
En el caso que reportamos, el paciente cursó con prácticamente todas las fases, excepto la cuarta. Las fases más notorias fueron la primera, caracterizada por acidosis metabólica severa y aumento de la brecha aniónica y la tercera, la insuficiencia renal aguda, que ameritó la administración de terapia de reemplazo renal.
Hallazgos de laboratorio
Las mediciones séricas o plasmáticas de las concentraciones de etilenglicol pueden proveer confirmación de la exposición tóxica de este agente. Sin embargo, los ensayos cuantitativos para esta sustancia no están disponibles en muchos laboratorios de los hospitales. Cuando las muestras son enviadas a centros de referencia puede resultar en retardo para establecer el diagnóstico y, por ende, se convierte en un retraso potencial en el tratamiento que puede salvar la vida, y en caso de disponer de ellos, la interpretación debe hacerse de manera cuidadosa. En los casos en los que hay una historia creíble de ingestión de etilenglicol, las muestras de laboratorio disponibles en muchos hospitales pueden proveer evidencia importante corroborativa de la exposición y también para ayudar a evaluar los metabolitos tóxicos que han iniciado a acumularse y cuáles modalidades de tratamiento están disponibles. En los casos en quienes la historia no está disponible o es incompleta, estas pruebas de escrutinio son más importantes, particularmente si las pruebas no están disponibles.1,15
Existe una pequeña correlación entre las concentraciones séricas de etilenglicol y la severidad del envenenamiento, lo que en ocasiones hace incierto el diagnóstico.4 Se ha reportado muerte en pacientes con concentraciones prácticamente indetectables de etilenglicol. La discrepancia subraya la importancia del diagnóstico temprano y el tratamiento de la intoxicación. La escasa correlación entre las concentraciones de etilenglicol y el resultado clínico es el resultado de la rápida depuración del componente original y la conversión a metabolitos tóxicos. El periodo de ventana durante el cual el etilenglicol puede detectarse puede ser relativamente estrecho.17
Se han desarrollado varios métodos para cuantificar las concentraciones de etilenglicol en suero o plasma. Existen métodos colorimétricos o ensayos enzimáticos, pero tienen limitacio nes respecto a la especificidad. Los ensayos colorimétricos están disponibles recientemente de manera comercial en Estados Unidos, pero sólo para uso veterinario, dando resultados cualitativos en sólo 30 minutos. En un estudio donde se usó la prueba colorimétrica, los autores concluyeron que esa prueba es sensible y especifica en casos de intoxicación por etilenglicol en humanos.22
La cromatografía de gas, usada para la medición del metanol, no puede utilizarse para analizar etilenglicol por su alto punto de ebullición, alta polaridad y baja presión de vapor, estos incon venientes pueden ser evadidos si se realiza una reacción química preliminar para transformar cualquier muestra de etilenglicol en un derivado más volátil y menos polar. Estas técnicas representan los últimos adelantos en los métodos usados para determinar las concentraciones de etilenglicol y de ácidos volátiles. Aunque estos métodos pueden proveer resultados precisos, el equipo es caro, son difíciles de interpretar por la instrumentación especial y habilidad requerida. Se han reportado, por ejemplo, falsos positivos en pacientes con errores innatos del metabolismo y en pacientes con concentraciones no tóxicas de otros glioles, como 2-3-butanediol y propilenglicol. Cuando las concentraciones del compuesto original son relativamente bajas, puede haber concentraciones potencialmente letales de los metabolitos circulantes tóxicos; así, se recomienda la medición plasmática del glicolato, que provee información acerca del nivel de intoxicación. Empero, la medición de glicolato es menos probable que esté disponible que las mediciones del etilenglicol, especialmente en casos urgentes.1,15,23
Diagnóstico
Debe sospecharse en los pacientes con estado mental alterado, acidosis metabólica severa con brecha aniónica y osmolar alta, hipocalcemia y cristaluria de oxalato de calcio.4,6,7
Pruebas de escrutinio de laboratorio
La baja masa molecular del etilenglicol, junto con su baja toxicidad intrínseca, por lo menos para las moléculas no transformadas, permite la existencia de grandes cantidades molares de estas sustancias en la circulación en el periodo inmediato después de la ingestión y de la absorción gastrointestinal. En esos casos, la osmolaridad plasmática se incrementa en proporción a la concentración de etilenglicol. La medición de la osmolaridad sérica representa una clave para la ingestión de esta sustancia, si el resultado es anormalmente alto y no explicado por algunos otros trastornos.1 La osmolaridad sérica es de 270-290 mOsm/kg H2O en un individuo sano. La diferencia entre la osmolaridad medida y la calculada, la brecha osmolar, resulta de la existencia de otros solutos en el suero. El incremento en la brecha osmolar generalmente se considera importante cuando es mayor de 10 a 15 mOsm/kg, lo que sugiere la existencia de sustancias de baja masa molecular que logran concentraciones séricas apreciables.1,23 Puede estar elevada en la primera hora de la ingestión y es el resultado de la existencia del etilenglicol por sí mismo, no por sus metabolitos tóxicos en el suero.5 La contribución del etilenglicol o del etanol puede calcularse de la siguiente manera, cada 16 mmol/L (100 mg/dL) de incremento en la concentración de etilenglicol contribuye aproximadamente 16 mosm/kg H2O y cada 22 mmol (100 mg/dL) de etanol contribuye con 22 mOsm/kg a la brecha osmolar.23 A medida que se metaboliza el etilenglicol, sus concentraciones plasmáticas disminuyen y entonces su contribución a la osmolaridad y a la brecha osmolar también declina; puede no ser aparente tardíamente en el curso del envenenamiento, encontrándose concentraciones letales de metabolitos tóxicos circulantes. Estos metabolitos en su forma neutral y en su forma aniónica tienen actividad osmótica; sin embargo, en su forma aniónica desplazan a los aniones bicarbonato activos y así los aniones ácidos no tienen efecto neto en la osmolaridad. Los ácidos orgánicos acumulados pueden ser detectados por sus efectos asociados en la brecha aniónica y el pH arterial. En la intoxicación por etilenglicol, hay correlación entre el glicolato plasmático (el principal anión ácido), el pH arterial y la brecha aniónica sérica. Cuando sobreviene frecuentemente es severa y la brecha aniónica está incrementada, como ocurrió en nuestro paciente, que padeció acidosis metabólica con pH inicial de 6.9 y con brecha aniónica de 35.79. Asimismo, la acidosis metabólica puede estar ausente y la brecha aniónica completamente normal aun cuando el paciente haya ingerido grandes cantidades de etilenglicol, esto puede deberse a que el paciente se presenta de manera temprana después de la ingestión y no ha habido tiempo suficiente para la transformación a metabolitos ácidos, aunque se han señalado otros factores, entre ellos el equilibrio electrolítico basal individual, la velocidad a la que se metaboliza el etilenglicol y la existencia o ausencia de un inhibidor enzimático semejante al etanol que puede retardar el metabolismo del etilenglicol.1,24 Se requieren estudios para confirmar los mecanismos específicos por los que ocurre acidosis metabólica de brecha aniónica normal en el contexto de la intoxicación por etilenglicol. Los clínicos deben, no obstante, estar alertas del potencial de la acidosis metabólica de brecha aniónica normal en pacientes con toxicidad significativa por etilenglicol y este diagnóstico no debe excluirse si la acidosis metabólica de brecha aniónica normal forma parte del trastorno metabólico en la presentación.24 Por estas razones, ni la brecha osmolar ni la brecha aniónica están universalmente presentes en los casos de intoxicación por etilenglicol y su ausencia, entonces, no puede usarse para descartar la toxicidad por etilenglicol. Al contrario, la coexistencia de acidosis metabólica con brecha aniónica y osmolar elevadas, aunque altamente sugerente de envenenamiento por etilenglicol, no es especifica de este cuadro.23
Cristaluria
En la intoxicación por etilenglicol, existen series y reportes de casos que con frecuencia documentan la existencia de cristales de oxalato de calcio en el análisis de orina. En el contexto adecuado, este hallazgo lleva al diagnóstico, como lo reportaron Luqman y colaboradores, en un paciente que llegó a urgencias obnubilado, con acidosis metabólica e insuficiencia renal; el examen de orina mostró numerosos cristales, que al ser examinados con luz polarizada mostraron ser birrefringentes, característica de los cristales de monohidrato de calcio.25 De igual manera Huhn y Rosenberg reportaron el caso de un paciente que se intoxicó por etilenglicol, se basaron en el hallazgo de acidosis metabólica no láctica persistente, cristaluria de oxalato de calcio y bajas concentraciones de calcio ionizado. El diagnóstico se confirmó posteriormente por la detección de trazas de etilenglicol y grandes cantidades de su metabolito, ácido oxálico, en el suero del paciente. Estos autores ilustran la forma de agujas de los cristales de monohidrato de calcio y lo estable cen como clave para establecer el diagnóstico de envenenamiento por etilenglicol. Cuando se investiga la acidosis metabólica no explicada en el departamento de Urgencias, la orina debe someterse a análisis microscópico en búsqueda de cristales de monohidrato o de dihidrato de oxalato de calcio.23,26 No obstante, pueden ocurrir como falsos positivos y falsos negativos. La cristaluria de oxalato de calcio se observa en pacientes con algunos trastornos intestinales y en pacientes con un trastorno poco frecuente, hiperoxaluria primaria. Los individuos sanos pueden padecer hiperoxaluria con o sin formación de cristales después de la ingestión de alimentos ricos en oxalato o después de la administración de grandes cantidades de ácido ascórbico. En contraste, la ausencia de cristaluria de oxalato no excluye la intoxicación por etilenglicol, como se ha reportado en algunos casos. El urianálisis falla para demostrar cristales de oxalato de calcio, pero el examen histopatológico muestra depósito marcado de oxalato de calcio en los riñones, en los túbulos renales y el intersticio. La ausencia de cristales puede deberse al tiempo en que se obtiene la muestra; los pacientes que acuden de manera temprana después de la ingesta y los que reciben terapia para prevenir la ruptura del glicol pueden no manifestar cristaluria nunca. Asimismo, los cristales pueden ser pasados por alto o no reportados en el examen de orina o pueden identificarse erróneamente como otros cristales diferentes del oxalato de calcio. Los cristales de oxalato de calcio en la orina son pleomórficos, abigarrados y birrefringentes cuando son vistos a través de la luz polarizada. En los casos de intoxicación por etilenglicol, el oxalato de calcio puede excretarse no sólo como cristales dihidratados, que son de forma curva (piramidales, octaédricos). Los cristales de monohidrato de calcio se aprecian en forma de agujas o de diferentes morfologías como cristales individuales aislados o agrupados en agregados. Los cristales de ácido hipúrico se han reportado en pacientes intoxicados con etilenglicol, tal vez representan malinterpretación, ya que las formas hexagonales elongadas y las que tienen forma de agujas corresponden a cristales de monohidrato de calcio y se confunden fácilmente con cristales de ácido hipúrico. Este ácido es un constituyente normal de muchos mamíferos herbívoros; el compuesto se halló inicialmente en la orina de los caballos, de ahí el nombre. El ácido hipúrico también puede estar presente en la orina humana, en ese caso, es derivado del aminoácido glicina en presencia de ácido benzoico; éste a su vez es un producto metabólico del tolueno. En teoría, la cristaluria de ácido hipúrico puede formarse en el enve nenamiento con etilenglicol en humanos, por una reacción de transaminación que involucra el ácido glioxílico para formar glicina, por la conjugación de glicina a ácido benzoico. La piridoxina se requiere en la primera reacción de la secuencia, pero, por ser un cofactor, no es consumida por la reacción. Por otro lado, la reacción de conjugación requiere una fuente continua de ácido benzoico o de su anión ben zoato. En teoría, las altas concentraciones de ácido hipúrico pueden ocurrir acompañadas de cristaluria; sin embargo, la evidencia documental es insuficiente. Algunos autores encontraron cristales en un paciente intoxicado con etilenglicol, parecidos a los del hipurato, pero al usar cristalografía radiográfica, un método definitivo de identificación de cristales, el autor mostró que los cristales fueron de monohidrato de oxalato de calcio.1 Si el resultado de pruebas de laboratorio más definitivas no está disponi ble, la detección de cristaluria de oxalato de calcio, particularmente las formas monohidratadas, proveen evidencia suficiente para el diagnóstico de la intoxicación por etilenglicol.23 El examen de la orina con lámpara de Wood puede ser de utilidad, porque los anticongelantes contienen fluoresceína.4 En 1990, Winter y colaboradores27 reportaron sus hallazgos de una prueba diseñada para demostrar si la fluoresceína incluida en muchas formulaciones de anticongelantes automotrices podría detectarse en la orina humana al ser expuesta a la luz UV. Ciertos fármacos y vitaminas o sus metabolitos poseen fluoresceína intrínseca (carbamazepina, niacina, caroteno y benzodiacepinas), lo que puede conducir a falsos positivos si estas sustancias han sido ingeridas y aparecen en la orina. Los falsos negativos pueden ocurrir si la muestra se obtiene unas pocas horas después de la ingestión o si el anticongelante tiene una formulación que no contiene fluoresceína. Esta prueba se usó subsecuentemente por lo menos como un sustituto para la identificación del anticongelante ingerido, en la orina, ropa, piel o en el contenido gástrico. Casavant y colegas28 estudiaron a 16 niños sanos y 30 hospitalizados por otras razones diferentes a las de intoxicación y concluyeron que la fluoresceína no es indicador de ingestión de anticongelantes en niños y recomendaron que esta prueba fuera abandonada; a estas mismas conclusiones llegaron el equipo de Wallace29 y el de Parsa.30 Con base en la información disponible en la actualidad se sugiere que la determinación de fluoresceína en la orina no se considere prue ba de escrutinio ante la sospecha de ingestión de anticongelantes.1 Pomara y colaboradores reportaron un caso fatal de intoxicación por etilenglicol en el que usaron microscopia de exploración láser en biopsia de riñón y eviden ciaron cristales de oxalato de calcio en forma de monohidrato y de dihidrato.10
Concentraciones sanguíneas de lactato
Las concentraciones incrementadas de lactato se han descrito en la intoxicación por etilenglicol, empero, de manera inconsistente. Muchas de las reacciones de deshidrogenización implicadas en el metabolismo del etilenglicol requieren el cofactor enzimático NAD+ produciendo la versión reducida NADH+. El exceso de NADH+ es reoxidado a NAD por el sistema intramitocondrial electrónico de electrones. Esta regeneración puede ser impedida si el ATP adicional no es requerido por la célula o si las moléculas tóxi cas interfieren con la operación del sistema de transporte o si las reacciones de la vía metabólica van contracorriente. En estas circunstancias la transformación de piruvato a lactato puede servir como una forma de reponer NAD+. En reposo, las concentraciones de lactato en el cuerpo están normalmente en equilibrio constante con el intermediario glicolítico piruvato, producido por el metabolismo de la glucosa. La conversión de piruvato a lactato está catalizada por la enzima lactato deshidrogenasa y requiere NADH+. Verelst y colegas expresaron que la acumulación de ácido láctico se debe a la depleción de la forma oxidada de la nicotinamida adenina dinucleótido y a la inhibición del metabolismo del piruvato por los metabolitos aldehídos, que pueden contribuir también al incremento de la brecha aniónica.31
Así, puede esperarse que la acumulación en exceso del NADH+ del metabolismo de grandes cantidades de etilenglicol exceda la tasa NAD+ a NADH+, tendiendo a llevar esta reacción en dirección al lactato e incrementando las concentraciones sanguíneas de lactato al repletar el NAD+. Sin embargo, las concentraciones sanguíneas inconsistentes de lactato en reportes de casos de estas ingestiones, incluidos aquéllos en los que se generaron grandes cantidades de metabolitos tóxicos, van en contra de este mecanismo como una fuente apreciable de la generación de lactato. En los pacientes con crisis convulsivas y en el periodo postictal, la acidosis y la acidosis láctica son esperadas debido al desequilibrio entre el aporte y la demanda de oxígeno tisular. Además, los modelos experimen tales de intoxicación por etilenglicol en ratas, perros y monos, así como algunos reportes de casos, no han mostrado incremento sustancial en el lactato. Debido a los efectos potenciales severos cardiopulmonares que pueden ocurrir en esta intoxicación, un mecanismo común de la hiperlactatemia asociada es el metabolismo anaerobio de la insuficiencia circulatoria, hipoperfusión o hipoxia. Otro posible factor que contribuye es la producción disminuida de ATP aerobio, causado por inhibición de la respiración celular, que promueve la obtención de ATP anaerobio, vía glucólisis.1
En 1999, Morgan y colaboradores32 realizaron un estudio prospectivo in vivo para determinar si el glicolato, que es similar al lactato, puede causar elevación artificial de las concentraciones medidas de L-lactato después de que algunas observaciones mostraron discrepancia en las mediciones plasmáticas de L-lactato en un paciente con intoxicación por etilenglicol y acidosis metabólica severa. Los autores hallaron que la acidosis en la intoxicación por etilenglicol se atribuyó primariamente a la formación de ácido glicólico. Además, los datos mostraron de manera concluyente que con los dos analizadores de gas usados es posible identificar incorrectamente el ácido glicólico como ácido láctico, más probablemente debido a la especificidad incompleta de la enzima L-lactato oxidasa en ambos modelos de analizadores de gases sanguíneos. El diagnóstico erróneo de la acidosis glicólica como acidosis láctica cuando se trata a un paciente con intoxica ción por etilenglicol puede retrasar el diagnóstico. Shirey y colaboradores33 llegaron a la misma conclusión, los autores concluyeron, además, que la medición de una brecha láctica usando dos diferentes tecnologías, una de las cuales es sensible al glicolato, puede ser suficiente en términos clínicos para hacer el diagnóstico de intoxicación severa por etilenglicol en el departamento de urgencias o en el marco de otros cuidados críticos. Porter y colegas advirtieron acerca del uso del método del lactato de Beckman Synchron en el que también lecturas falsas positivas de acidosis láctica se detectaron en casos de intoxicación por etilenglicol.15 Woo y colaboradores34 confirmaron que la interferencia con glicolato en la medición del lactato es variable, dependiendo del método y de la plataforma analítica usada por el laboratorio. Brindley y su grupo35 realizaron un estudio en el que varias concentraciones de metabolitos del etilenglicol se adicionaron a una muestra de sangre venosa y probada con cinco diferentes analizadores de lactato comunes. Las muestras evidenciaron concentraciones marcadamente elevadas de lactato cuando se adicionó glicolato y también cuando se agregó glioxilato. Los autores concluyeron que los resultados falsos positivos se debieron a la reacción cruzada en tre los metabolitos del etilenglicol y la L-lactato deshidrogenasa.31,36
Medición por laboratorio de los metabolitos del etilenglicol
Debido a que la toxicidad del etilenglicol depende de su oxidación a ácidos orgánicos, varios otros métodos se han usado para medir estos productos. El ácido glicólico, el metabolito predominante, se ha medido por cromatografía de gases, por método colorimétrico y por isota-cofóresis.23 Cuando las pruebas están disponibles pueden tardar entre cuatro y seis horas, y son caras. Puede utilizarse la brecha osmolar con un estimado de la concentración de etilenglicol, multiplicando el exceso de la brecha osmolar por 6.2 ([brecha osmolar - 10] x 6.2).
Otras alteraciones de laboratorio
La intoxicación por etilenglicol puede resultar en varias otras alteraciones de laboratorio, además de la acidosis metabólica, la brecha aniónica aumentada y la brecha osmolar. La hipocalcemia está descrita en muchos reportes de casos de intoxicación por etilenglicol, generalmente se asume que el depósito tisular de calcio indisoluble es la causa. Sin embargo, no hay datos actuales que apoyen este concepto. La disregulación de la hormona paratiroidea también puede contribuir potencialmente a la hipocalcemia. La actividad aumentada de la amilasa en el plasma causada por pancreatitis o por inflamación de las glándulas salivales también se ha reportado. La azoemia sobreviene durante la tercera fase clásica de la intoxicación. Se ha descrito también trombocitopenia.1,6,15
Tratamiento
La severidad de la intoxicación por etilenglicol varía en un amplio espectro según la dosis ingerida, el tiempo transcurrido entre la ingestión y la atención médica, el consumo concomitante de alcohol y otros factores. Si el paciente acude tardíamente después de la ingestión de grandes cantidades, el paciente puede estar en disfunción orgánica múltiple. El tratamiento de apoyo incluye las medidas que son válidas para pacientes severamente intoxicados o críticamente enfermos en general. La clave del tratamiento es la administración de antídotos farmacológicos específicos para retardar la aparición de metabolitos tóxicos y permitir la eliminación del compuesto original y de los productos tóxicos. Hay dos formas primarias disponibles de antídotos, el etanol y el fomepizol. Estos tratamientos farmacológicos actúan por mecanismos similares y pueden ser igualmente efectivos si se administran de manera apropiada. Deben considerarse formas de tratamiento mutuamente excluyentes.1
Las indicaciones para iniciar tratamiento con etanol o fomepizol son las siguientes: a) concentraciones séricas de etilenglicol mayores de 20 mg/dL; b) historia documentada de ingestión reciente de cantidades tóxicas de etilenglicol en conjunto con brecha osmolar mayor de 10 mOsm/kg, o c) sospecha clínica de etilenglicol, en conjunto, con al menos dos de los siguientes: 1) pH arterial menor de 7.3; 2) bicarbonato sérico o CO2 menor de 20 mmol/L; 3) brecha sérica osmolar mayor de 10 mOsm/kg; 4) cristaluria de oxalato de calcio. En tanto, las indicaciones de hemodiálisis son: a) deterioro del estado clíni co a pesar de terapia de apoyo; b) lesión renal aguda con creatinina sérica mayor de 3.0 mg o incremento en la creatinina sérica de 1 mg/dL; c) alteraciones del equilibrio ácido base que no responden al tratamiento estándar (acidosis metabólica).1,4,6,7,15
Medidas de apoyo y descontaminación digestiva
La terapia inicial siempre debe iniciar con garantizar la vía aérea del paciente, el mantenimiento de la ventilación y circulación. La intubación orotraqueal puede ser necesaria en casos tardíos en los que el paciente está en coma o tiene actividad convulsiva, como ocurrió en nuestro paciente. Si la medición de glucosa no está disponible en el primer minuto de llegada, debe administrarse empíricamente un bolo IV de glucosa. La inducción del vómito está contraindicada, aun en el paciente consciente debido al riesgo de depresión sensorial. El lavado gástrico puede ser una consideración en la ingestión de grandes cantidades, siempre y cuando el paciente acuda una hora después de la ingestión y tenga colocado el tubo endotraqueal.1,2,5,7 Hay datos limitados acerca del uso del carbón activado, pero es probable que sea útil cuando se sospecha la ingestión de polifarmacia.7 Las convulsiones se tratan con fármacos parenterales anticonvulsivos. Deben tomarse muestras para la medición de las concentraciones de electró litos séricos, incluidos calcio, brecha aniónica, osmolaridad medida, nitrógeno ureico, glucosa, análisis de gases arteriales y examen de orina. Las concentraciones de amilasa, lipasa y creatincinasa también deben evaluarse.
Bicarbonato de sodio
La acidosis de la intoxicación por etilenglicol puede ser profunda y pueden requerirse grandes cantidades de bicarbonato. Existen muchos reportes en los que el pH varía de 6.3 a 6.9. Tales grados impresionantes de acidemia son razones para la administración de álcali, pero también hay razones teóricas para la corrección de la acidosis. Debido a que los metabolitos son principalmente ácidos carboxílicos y que muchos de los efectos adversos afectan al sistema nervioso central, cualquier medida encaminada a disminuir las moléculas tóxicas al sistema nervioso central es razonable.
En estudios animales se ha demostrado beneficio con la administración de bicarbonato. En un modelo de intoxicación por etilenglicol en ratas, Borden y colaboradores37 reportaron una tasa de 71% de supervivencia en las tratadas con bicarbonato comparadas contra 14% de las que no lo recibieron. La cristalización cortical también disminuyó, con incidencia de 94% en las no tratadas contra 55% de las que recibieron bicarbonato. Así, las guías de la Academia Americana de Toxicología recomiendan la administración de bicarbonato en los pacientes intoxicados con etilenglicol si el pH arterial es menor de 7.30.
Bloqueo de la oxidación del etilenglicol
La oxidación del etilenglicol por la alcohol deshidrogenasa es un paso crítico en su conversión a oxalato, que produce acidosis metabólica y posteriormente insuficiencia renal. Así, si la oxidación de etilenglicol a oxalato puede bloquearse, puede prevenirse la toxicidad fatal debido a la ingestión de este compuesto.6
Etanol
Los estudios detallados de la oxidación enzimática del etilenglicol demostraron que el alcohol etílico, el sustrato natural, es un potente inhibidor de la oxidación del etilenglicol. Entonces, las bajas concentraciones de alcohol etílico previenen la oxidación de grandes cantidades de etilenglicol.
Por muchos años el etanol ha sido el antídoto universal contra la intoxicación por etilenglicol. Se ha administrado exitosamente por vía IV y oral. La afinidad y la eficacia catalítica de la alcohol deshidrogenasa son más altas para el etanol que para el etilenglicol, cerca de 65 veces más por el etanol. En presencia de etanol, esta enzima, en efecto, cataliza preferentemente la ruptura del etanol, inhibiendo funcionalmente la actividad de la enzima, mucho más que por la oxidación del etilenglicol.
La eficacia del etanol como inhibidor competitivo de la alcohol deshidrogenasa se ha demostrado en modelos animales. Por ejemplo, ratas intoxicadas con 10 mL/kg de etilenglicol y tratadas con etanol a 0.4% g/kg cada 6 horas por 6 dosis tuvieron supervivencia de 73% com paradas contra 14% de las que no recibieron tratamiento. La supervivencia se incrementó a 89% si el tratamiento consistió de bicarbonato y etanol. La formación de cristales de oxalato de calcio disminuyó de 94% en el grupo control a 25% en los animales tratados con etanol, mientras que no hubo cristalización en los animales tratados con la combinación de etanol y bicarbonato.
Una concentración sérica de etanol de 100-150 mg/dL (21.7-32.6 mmol/dL) es generalmente adecuada para la inhibición competitiva del metabolismo del etilenglicol, incluso inhibiéndola completamente. El etanol puede mezclarse con agua y se difunde en todos los compartimientos líquidos del cuerpo, con un volumen de distribución de cerca de 0.68 L/kg en hombres y de 0.63 L/kg en mujeres. La dosis de carga convencional recomendada para conseguir rápidamente una concentración de etanol en el intervalo descrito es de 0.6 g de etanol por kg de peso corporal, equivalente a 0.76 L de etanol absoluto (al 100% de etanol anhídro) por kg de peso corporal. La carga puede conseguirse por administración IV de una solución de etanol a 10% en agua estéril. Esta dosis asume que la concentración de etanol del paciente es de 0 al iniciar el tratamiento. La concentración basal de etanol debe evaluarse porque algunos pacientes tienen etanol en la circulación al haber consumido etanol junto con el etilenglicol. No se requiere dosis de carga si las concentraciones basales de etanol están en el intervalo o por encima del mismo. En pacientes con concentraciones detectables pero bajas, se administra una dosis de carga proporcionalmente menor.
La experiencia clínica y los datos obtenidos en voluntarios sanos sugieren que esta dosis de carga, que está basada en datos limitados de pacientes, comúnmente falla para conseguir el objetivo de la concentración sérica de etanol, especialmente cuando se administra por vía oral. Como resultado, algunas fuentes aconsejan rutinariamente una dosis de 0.7 g/kg en lugar de 0.6 g/kg. También se han recomendado dosis de carga de 0.8 g/kg.
Una vez que la dosis de etanol está en curso, es necesario mantener la concentración de etanol en el intervalo objetivo. La dosis requerida de etanol para mantener la concentración de etanol en el intervalo deseado es, en promedio, de aproximadamente 66 mg/kg/h en individuos no habituados al etanol (0.83 mL de etanol a 10%) y de 154 mg/kg/h en los usuarios crónicos (1.96 mL de etanol a 10%). La dosis debe titularse por mediciones de etanol sérico cada una a dos horas; una vez que se ha alcanzado la dosis para el mantenimiento, los ensayos pueden requerirse cada dos a tres horas. Los estudios deben hacerse cada una o dos horas si la concentración de etanol está fuera de los intervalos terapéuticos, si la dosis es interrumpida o si se requiere diálisis. Cuando se administra etanol a 5% las tasas de infusión deben ser de 1.66 y 3.92 mL/kg/h. Pueden requerirse dosis más altas que las citadas, particularmente en alcohólicos crónicos que están habituados a ingerir grandes cantidades de alcohol.
Cuando no se dispone de etanol farmacéutico, pueden administrarse diversos licores por vía oral, puede darse whisky comercial o algún etanol de grado farmacéutico apto. Esta ruta no está permitida en pacientes con sensorio alterado por el riesgo de broncoaspiración si sobreviene el vómito. Tampoco se recomienda en pacientes que no están acostumbrados a ingerir este tipo de licor por la probabilidad de vómito, aun si la preparación está diluida. La dosis de carga se calcula de la siguiente manera: 200/grados [de la bebida administrada] mL/kg/h, mientras que la dosis de sostén se obtiene de la siguiente forma: 30/grados [de la bebida] mL/kg/h. Si se dispone de etanol a 43% la dosis de carga es de 1.8-2.1 mL/kg (promedio 1.5 mL/kg de peso corporal), en no bebedores la dosis de mantenimiento es de 0.2 mL/kg/h; cuando se prescribe hemodiálisis la dosis es de 0.5 mL/kg/h. En los bebedores crónicos es de 0.46 mL/kg/h y durante la hemodiálisis es de 0.77 mL/kg/h.38,39 Este in cremento se requiere para compensar el etanol removido durante la hemodiálisis, en algunos casos se requiere que la dosis sea duplicada o triplicada, pero el incremento es afectado por diversos factores y es difícil de predecir. La dosis típica de etanol durante la hemodiálisis es de 169 mg/kg/h en términos de etanol absoluto. Esta dosis es equivalente a 2.13 mL/kg/h si se administra una dosis de etanol parenteral a 10%. Para pacientes con consumo regular de etanol, una dosis representativa en hemodiálisis es de 257 mg/kg/h, equivalente a 3.26 mL/kg/h.1
La absorción enteral del etanol está escasamente definida y depende de múltiples factores. Gran parte de absorción ocurre en el estómago (20%) y en el duodeno (80%). Hay un metabolismo significativo de primer paso del etanol liberado vía enteral antes de que se distribuya a través del cuerpo, debido principalmente al paso directo, vía del sistema venoso portal al hígado, pero también debido a la presencia de alcohol deshidrogenasa en la mucosa gástrica. Este nivel depende de la raza (alto en caucásicos), género (alto en hombres) y por el consumo concomitante de fármacos (reducido en aspirina y por antagonistas de los receptores H2). El metabolismo del etanol en la mucosa es más pronunciado con el vaciamiento gástrico retardado, que puede estar relacionado con la dieta (alimentos grasos), fármacos procinéticos y enfermedades críticas. Tal efecto no es visto cuando el etanol es entregado directamente al duodeno o más allá, como lo reportaron Holyoak y colaboradores.7 El metabolismo del primer paso del etanol tiene importantes ramificaciones para las concentraciones tisulares y séricas del etanol absorbido vía enteral, lo que resulta en concentraciones bajas que pueden ser vistas con la administración endovenosa. En el caso de la intoxicación por etilenglicol, la diferencia puede no ser importante, la finalidad de la administración de etanol es saturar la alcohol deshidrogenasa hepática, esto puede ocurrir vía la ruta enteral, debido a la entrega directa portal, aunque el significado de esta diferencia es probablemente pequeño.
Otros factores que influyen en la concentración sérica de etanol y, por tanto, en la saturación de la alcohol deshidrogenasa hepática, incluyen su distribución tisular, rutas alternas de metabolismo y el flujo sanguíneo hepático. El metabolismo hepático del etanol no siempre sigue una cinética de orden cero, ya que no es metabolizado sólo por la alcohol deshidrogenasa. En el consumo crónico de etanol, la inducción de las enzimas del citocromo P450 permite una vía metabólica alterna con una tasa alta de Michaelis, que resulta en un metabolismo más rápido del etanol y bajas concentraciones séricas. Además, la distribución del etanol a través del cuerpo se relaciona con la cantidad de agua corporal presente y, por tanto, es un factor de la edad, género y masa corporal magra. Esta distribución y su resultante equilibrio con el volumen sanguíneo circulante no es constante, siendo alterados por la perfusión tisular, con cambios rápidos en las concentraciones séricas. También el flujo sanguíneo en los vasos esplácnicos puede estar alterado debido a los estados de ayuno, ejercicio y enfermedades críticas que resultan en perfusión hepática alterada y, por ende, en el metabolismo del etanol. Holyoak y colaboradores concluyeron que cuando el etanol endovenoso no está disponible, puede sustituirse con etanol enteral con buenos resultados.7
La clave para mantener la dosis efectiva de etanol es obtener mediciones frecuentes y usar la información para titular la dosis. De manera típica, las concentraciones de etanol deben medirse cada hora de manera inicial y durante la hemodiálisis, y cada dos horas cuando el paciente esté fuera de diálisis, una vez que la concentración de etanol sea estable. La infusión de etanol es titulada para mantener la concentración entre 100 y 150 mg/ dL. No son necesarias concentraciones más altas, pueden causar sedación innecesaria, ebriedad y pérdida de los reflejos protectores. Deben evitarse las concentraciones bajas porque pueden ser insu ficientes para inhibir completamente la conversión enzimática. En nuestro caso, la administración de etanol se prescribió con dosis menores a lo recomendado, sin tener, además, mediciones de las concentraciones séricas del mismo para hacer ajustes antes ni durante la hemodiálisis.
Aunque las soluciones parenterales de etanol tienen regulaciones aprobadas para uso clínico por la Dirección de Alimentos y Fármacos de Estados Unidos, no están aprobadas para admi nistrarse como tratamiento de la intoxicación por etilenglicol. A pesar de ello, el etanol tiene una larga historia de uso clínico para este propósito. Modelos animales, reportes de casos y series de casos atestiguan la efectividad del tratamiento con etanol administrado, ya sea vía endovenosa u oral, pero los ensayos clínicos controlados con humanos son insuficientes. Existen desventajas conocidas del tratamiento con etanol, tiene efectos sedantes, además de la dosis, cuyo cálculo puede ser complejo y no familiar para algunos clínicos, debido sobre todo a la infrecuente ocasión para su prescripción. La administración oral conlleva potenciales efectos secundarios adicionales, que incluyen dolor abdominal, náuseas, gastritis, sangrado gastrointestinal, vómitos y neumonitis por aspiración.
La administración de etanol debe continuarse hasta que las concentraciones de etilenglicol sean indetectables o alternativamente hasta que las concentraciones sean menores de 20 mg/dL y el pH del paciente sea normal.1,4,15
En un estudio efectuado en Ottawa, Canadá, con seguimiento de nueve años, Wedge y colaboradores concluyeron que la administración de etanol no sólo es difícil de titular, sino que está asociada con efectos adversos, los efectos en el sistema nervioso central son los más frecuentes; los autores sugieren que deben tomarse en cuenta los efectos adversos cuando se elija el etanol como antídoto.40
Lepik y colaboradores, en un estudio realizado en 10 hospitales de cuatro áreas geográficas de Columbia Británica, Canadá, incluyeron 145 casos de intoxicación con etilenglicol tratados con etanol, hubo 305 errores en la medicación en 113 de los 145 casos, los errores fueron la duración inapropiada del tratamiento, dosis excesiva y vigilancia inadecuada. Además, documentaron errores peligrosos en 28 de los 145 pacientes, los más frecuentes fueron el retraso en el inicio del tratamiento y la dosis excesiva del antídoto. Si bien durante el periodo de estudio, los casos de errores en la medicación con etanol descendieron de 80% en los años 1996-2003 a 45% en 2005, se atribuyó a mayor consulta toxicológica. Con base en los resultados, para minimizar los errores en la medicación, los autores recomiendan las siguientes pautas: 1) reducir el riesgo de malinterpretar los estudios de laboratorio, 2) incorporar guías de tratamiento, 3) evitar el retardo en acceder al antídoto, 4) prevenir la interpretación errónea de las indicaciones médicas, 5) evitar errores de cálculo, 6) indicar claramente la dosis del antídoto antes, durante y después de la hemodiálisis y 7) evitar los errores de transcripción entre hospitales.41
Fomepizol
El fomepizol, 4 metilpirazole, es un inhibidor competitivo de la alcohol deshidrogenasa, que previene la formación de metabolitos del etilenglicol. Es más efectivo cuando se administra de manera temprana, antes de que se formen cantidades significativas de los metabolitos tóxicos. El pronóstico depende del tiempo transcurrido desde la ingestión hasta el momento en que se inicia el tratamiento y de la cantidad de metabolitos tóxicos que se han acumulado, más que de la concentración plasmática del compuesto original, al tiempo en que se administra el fomepizol. Fue aprobado por la FDA para el tratamiento de la intoxicación por etilenglicol en 1997.42 No tiene efectos hepatotóxicos como su antecesor, el pirazol. La afinidad por la alcohol deshidrogenasa es 8,000 veces superior a la del etanol.43
La evidencia primaria de la eficacia del fomepizol en humanos deriva de dos series retrospectivas de casos, de otra serie de casos relacionada con la toxicidad del metanol en Noruega y dos estudios prospectivos, estos últimos son subestudios del Methylpyrazole for Toxic Study Group, META. Éste enroló 23 pacientes consecutivos, de los que 19 cumplieron los criterios para intoxicación por etilenglicol. Dieciocho pacientes supervivieron; diez tuvieron función renal normal al momento de la presentación, sin mostrar lesión renal subsecuente a pesar de concentraciones tan altas de etilenglicol como de 446 mg/dL y valores de pH tan bajos como 7.16. La dosis inicial fue de 15 mg/kg por vía IV en 30 minutos, seguida de una dosis de mantenimiento de 10 mg/kg cada 12 horas, hasta alcanzar concentraciones de etilenglicol menores de 0.2 g/L. En caso de hemodiálisis, la dosis se incrementó en 1 a 1.5 mg/kg, ya que el fármaco es dializable. La con centración plasmática requerida para inhibir la alcohol deshidrogenasa es de 0.8 mg/mL. La vida media del etilenglicol sin hemodiálisis fue de 19.7 horas con aclaramiento plasmático entre 17 y 70 mL/min. El tratamiento de acuerdo con la Academia Americana de Toxicología se describe a continuación: dosis inicial si el paciente no está en hemodiálisis de 15 mg/kg en 30 minutos, seguido de 10 mg/kg en 30 minutos cada 12 horas por 4 dosis y posteriormente 15 mg/kg en 30 minutos cada 12 horas hasta que 1) las concentraciones de la toxina sean indetectables o 2) las concentraciones plasmáticas del tóxico sean menores de 20 mg/dL, el pH esté en intervalos normales y el paciente esté asintomático. Si el paciente es sometido a hemodiálisis y han pasado menos de 6 horas desde la última dosis, se pasa a la siguiente dosis; sin embargo, si han transcurrido más de 6 horas después de la última dosis se vuelve a administrar una dosis de carga de 15 mg/kg. Durante la hemodiálisis debe administrarse una dosis cada 4 horas en 30 minutos calculada a razón de 15 mg/kg. Al con cluir la hemodiálisis y si ha transcurrido menos de una hora de la última dosis no se administra una nueva dosis, pero si han pasado una a tres horas desde la última dosis, se administra una dosis de 7.5 mg/kg al final del procedimiento. Si ya transcurrieron más de tres horas se administra una nueva dosis de carga de 15 mg/kg. La dosis posdiálisis se administra 12 horas después de la última dosis a razón de 15 mg/kg en 30 minutos cada 12 horas, hasta que las concentraciones séricas del etilenglicol sean menores de 20 mg/ dL. A pesar de estas complejidades, el fomepizol es más fácil de administrar que el etanol, las con centraciones séricas no son requeridas porque el régimen de la dosis está predeterminado en lugar de empíricamente titulado. Estas ventajas, junto con el perfil de efectos secundarios del fomepizol, lo hacen preferible al etanol cuando está disponible.1,42,43 En el estudio de Lepik y su grupo, se reportaron errores en la medicación en 45% de los pacientes que recibieron fomepizol, mientras que ocurrieron en 113 de los 145 que recibieron etanol. Si bien, tiene un perfil de seguridad adecuado, no está libre de efectos secundarios. En el estudio META, se documentaron dos casos de bradicardia, dos episodios de cefalea y dos convulsiones en pacientes distintos, así como empeoramiento de la función renal en nueve pacientes (que ya tenían elevada la creatinina). Recientemente el grupo de Lepik reportó a un paciente que recibió tratamiento con fomepizol y que padeció bradicardia e hipotensión, concluyeron que estos efectos pueden haber sido precipitados por la hemodiálisis, lo que sugiere que los pacientes deben vigilarse durante y al finalizar la infusión.43
El grupo de estudio META describió también la toxicinética del etilenglicol en pacientes intoxicados que recibieron fomepizol como antídoto, hallaron que la vida media fue de cerca de 19 horas en pacientes con función renal normal, mientas que se alargó significativamente en pacientes con concentraciones de creatinina mayores de 130 mmol/L (1.47 mg/dL). En el caso reportado por Hoffman y colaboradores, el paciente tuvo una concentración de creatinina de 137 mmol/L y la eliminación fue marcadamente prolongada, lo que confirma que la creatinina sérica elevada predice la eliminación lenta del etilenglicol.8 Un estudio retrospectivo que se realizó en 40 pacientes, siguiendo un modelo toxicinético, encontró que la vida media fue de 14.2 horas; los autores concluyeron, además, que en la intoxicación con etilenglicol, los pacientes que no padecen insuficiencia renal o acidosis metabólica pueden beneficiarse con la administración de fomepizol como mono-terapia.44 Hay muchos reportes de casos de pacientes intoxicados con etilenglicol que fueron tratados exitosamente con fomepizol solo. La característica común de estos pacientes fue que mantuvieron función renal y pH normales, un ejemplo representativo fue el de un paciente de 61 años, con concentraciones séricas de 202 mg/dL, pH de 7.17 y creatinina de 0.9, tratado sin hemodiálisis; el paciente recibió tratamiento agresivo con líquidos, seis dosis de fomepizol y bicarbonato de sodio. Se demostró que aún con concentraciones mayores de 50 mg/dL de etilenglicol, la administración de fomepizol más cuidados de soporte fueron efectivos y obviaron la necesidad de hemodiálisis.45 El grupo de Buchanan reportó un caso con concentraciones séricas de etilenglicol de 700 mg/dL, función renal normal y acidosis metabólica leve que no se sometió a hemodiálisis; recibió tratamiento sólo con fomepizol, lo que corrobora que este antídoto puede administrarse solo, siempre y cuando la función renal esté intacta y no haya acidosis metabólica significativa.46
Hemodiálisis
La terapia con inhibidores no detiene completamente la generación de productos tóxicos del etilenglicol, pero disminuye sustancialmente su producción. Cuando el paciente tiene función renal normal, el etilenglicol también se excreta en la orina. Durante esta eliminación fisiológica, la existencia de concentraciones terapéuticas de etanol o fomepizol previene la formación excesiva y la acumulación de intermediarios tóxicos. Empero, ninguno de los dos tratamientos apresura la eliminación. El etilenglicol y sus metabolitos tóxicos son moléculas pequeñas y son dializables sin problemas. Por estas razones, el tratamiento de los pacientes intoxicados con etilenglicol ha incluido convencionalmente la hemodiálisis para remover el compuesto original y los metabolitos. Constituye, además, una intervención terapéutica vital en pacientes con suficiente atraso entre el tiempo de la ingestión y el inicio del tratamiento con los inhibidores de la alcohol deshidrogenasa, durante el cual ya hay concentraciones significativas de glicolato. Las indicaciones convencionales aconsejan hemodiálisis para todos los pacientes cuyas concentraciones séricas de etilenglicol sean superiores a 50 mg/dL; sin embargo, la evidencia más reciente y las recomendaciones de la Academia Americana de Toxicología sugieren que la diálisis no es necesaria por arriba de estas concentraciones, si alguno de los antídotos se está administrando y el paciente está asintomático, tiene pH normal y no hay ninguna otra indicación de hemodiálisis.1,15
El grupo español del Hospital Universitario La Paz, en Madrid, España, reportó 14 casos de intoxicación por alcoholes, incluidos dos con etilenglicol que fueron sometidos a hemodiálisis con membranas de alto flujo; concluyeron que el tratamiento debe iniciarse de manera temprana, con un dializador de alto flujo y gran superficie con lo que se obtienen mejores resultados, ya que elimina mayor cantidad de tóxico por hora.47 Pueden usarse también las técnicas continuas particularmente cuando la hemodiálisis cause inestabilidad hemodinámica, aunque esta modalidad es menos eficaz que la hemodiálisis convencional.48
Se ha descrito incremento en las concentraciones plasmáticas de etilenglicol entre 12 y 36 horas después de haber terminado la hemodiálisis en algunos casos, atribuido a la redistribución de los compartimientos tisulares al compartimiento plasmático, este fenómeno no es predecible y el incremento en las concentraciones puede ser significativo, requiriendo la continuación del etanol o fomepizol después de la diálisis con vigilancia de la osmolaridad sérica, electrólitos, brecha aniónica y brecha osmolar, con reevalua ción en intervalos de dos a cuatro horas por un periodo de 12 a 36 horas después de terminar el procedimiento,49 continuando con el inhibidor de nueva cuenta hasta que las concentraciones sean indetectables o alternativamente menores de 20 mg/dL, pH arterial normal, brecha aniónica normal y el paciente esté asintomático.
Terapia de apoyo
La piridoxina a dosis de 50 mg cada 6 horas puede inhibir el metabolismo del ácido glicólico a ácido oxálico al actuar como factor en el metabolismo del ácido glicólico a productos no tóxicos. La tiamina a dosis de 100 mg cada 6 horas está indicada para estimular la conversión de glioxalato a alfa-hidroxi-beta-cetoadipato, un metabolito no tóxico. Sin embargo, se carece de evidencia para recomendar la administración de manera rutinaria, pero debe indicarse en pacientes con riesgo de déficit de piridoxina y, por tanto, en pacientes que se sabe o se sospecha padecen alcoholismo crónico o desnutrición. La administración de calcio no está recomendada en los pacientes hipocalcémicos intoxicados con etilenglicol, a menos que haya manifestaciones clínicas; la administración puede incrementar la formación de cristales de oxalato de calcio con el consecuente depósito en el cerebro, los riñones, el corazón y otros órganos vitales.1,5
CONCLUSIONES
La intoxicación por etilenglicol es una urgencia médica, que de no ser tratada puede resultar fatal. Proponemos la administración de etanol por vía enteral para tratar esta intoxicación cuando no se disponga del tratamiento parenteral con alguno de los antídotos, con protección de la vía aérea y manteniendo al paciente en ventilación mecánica. La dosis debe ser la adecuada para evitar que el tratamiento sea ineficaz e idealmente valorarse con mediciones de las concentraciones séricas de etanol para guiar el tratamiento. Debe asociarse con hemodiálisis para la remoción de los metabolitos tóxicos cuando el paciente es llevado de manera tardía al servicio de urgencias y evidentemente en caso de insuficiencia renal aguda.

