











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkANTECEDENTES
La enfermedad cardiovascular aterosclerosa (ECV) es la primera causa de muerte en el mundo, por lo que su prevención es el mayor reto de salud pública. Para el año 2030 se prevé que haya más de 2.3 millones de casos, con un costo estimado de más de un millón de millones de dólares anuales, a menos que se tomen medidas urgentes enfocadas a reducir las cifras de colesterol de lipoproteínas de baja densidad (C-LDL),1 que son el origen de este padecimiento.
La molécula de LDL es esférica; su diámetro mide 220 nm y pesa aproximadamente 3,000 kDa.2 La partícula contiene 1,500 moléculas de ésteres de colesterol en un núcleo hidrofóbico, rodeado por una monocapa anfipática que incluye 800 moléculas de colesterol libre y 500 moléculas de fosfolípidos. Cada partícula de LDL tiene una sola copia de apolipoproteína B100 (APO B100, peso molecular 500 kDa), que conforma una sexta parte del peso total de la lipoproteína.3
Las moléculas de LDL son el producto final de la vía endógena de síntesis de colesterol. Ésta se inicia con la síntesis hepática de las lipoproteínas de muy baja densidad (VLDL), que paulatinamente pierden sus triglicéridos por acción de la lipasa de las lipoproteínas plasmáticas, y después por la lipasa hepática hasta convertirse en una molécula madura de LDL, prácticamente desprovista de triglicéridos.2
Las LDL envían el colesterol a la periferia; sin embargo, cuando su concentración se eleva pueden permear las paredes vasculares y sufrir en el espacio subendotelial la oxidación de sus moléculas, lo que activa un proceso inflamatorio e inmunológico en la pared arterial. De esto resulta el reclutamiento de células inmunocompetentes de estirpe mononuclear2 y la fagocitosis de las LDL oxidadas por los receptores basureros (scavenger) de los macrófagos, que se convierten en células espumosas, principal componente lipídico de la placa de ateroma.
Las estatinas son el tratamiento farmacológico de elección para reducir las concentraciones circulantes de LDL. Numerosos estudios de prevención primaria y secundaria, con distribución al azar y controlados con placebo han demostrado que estos medicamentos son relativamente seguros y con baja incidencia de efectos adversos.4 Las estatinas han permitido disminuir no sólo la concentración de C-LDL, sino también el riesgo cardiovascular (la reducción de 1 mmol de C-LDL, que equivale a 38 mg reduce 20% el riesgo cardiovascular).5
Aunque en general las estatinas son medicamentos bien tolerados, algunos pacientes (10-15%) sufren efectos adversos, principalmente mialgias que pueden convertirse en miopatías, o en raros casos rabdomiólisis que impiden su administración.5
El riesgo cardiovascular residual es el que persiste después de ingerir las dosis máximas toleradas de estatinas, solas o con otros medicamentos. Se debe, en primer lugar, a intolerancia a estatinas, a factores como la reducción insuficiente del C-LDL, el incremento de la lipoproteína a [Lp(a)] y triglicéridos, o cifras reducidas del colesterol unido a lipoproteínas de alta densidad (C-HDL). En este último caso se sabe que los pacientes con bajas concentraciones de HDL tienen mayor riesgo cardiovascular; sin embargo, hasta ahora no se ha podido demostrar el beneficio de medicamentos como los inhibidores de la proteína de transferencia de ésteres de colesterol (CETP), dirigidos a incrementar las HDL como objetivo principal del tratamiento (Figura 1).16
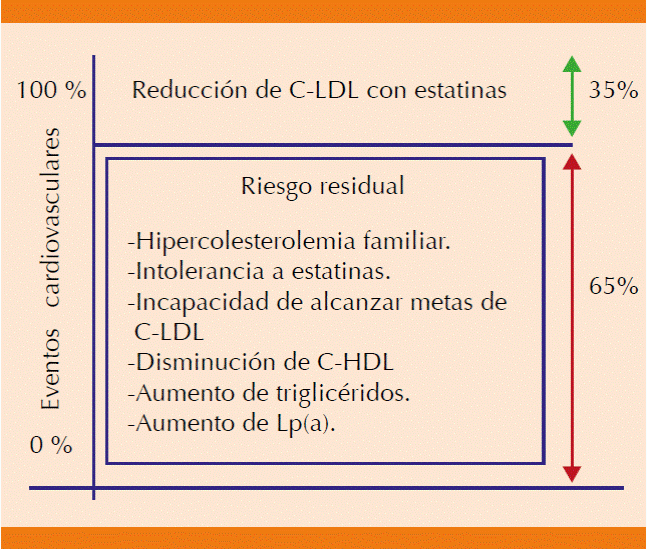
Figura 1 A pesar del tratamiento con estatinas, hay pacientes en alto riesgo de tener eventos cardiovasculares. Este riesgo está dado por factores no modificables, como la edad y el sexo, y potencialmente modificables. En conjunto, este riesgo se llama "riesgo cardiovascular residual". Tomada de la referencia 1.
La meta terapéutica en pacientes con enfermedad cardiovascular es alcanzar cifras de C-LDL <70 mg/dL;3,7 sin embargo, la eficacia de la monoterapia con estatinas es apenas de 37%. Ante este hecho, el clínico se enfrenta a la opción de incrementar la dosis hasta el máximo recomendado o prescribir estatinas de mayor potencia. Desafortunadamente, con ninguna de estas opciones se han alcanzado las metas terapéuticas.8 En cuanto a la adición de un segundo fármaco hipolipemiante, se ha observado que la combinación de estatinas y niacina ha aumentado la eficacia a 46%, pero esta última no ha mostrado beneficio cardiovascular y no es bien tolerada por los pacientes, mientras que las estatinas con fibratos disminuyen la efectividad a 35%.9,10 Estas combinaciones de fármacos elevan el costo y el riesgo de efectos colaterales. Hasta ahora cualquier monofármaco diferente a las estatinas tiene eficacia similar a la del placebo.11
Una de las combinaciones más prescritas es la de estatinas con 10 mg de ezetimiba (frecuentemente simvastatina y ezetimiba). Aunque el estudio Improve-it plantea la superioridad de esta combinación sobre las estatinas solas, el significado real de este estudio ha sido muy cuestionado y el costo de la combinación resulta muy elevado.2
Un número significativo de pacientes tienen riesgo cardiovascular alto a causa de formas primarias de hipercolesterolemia. Estas formas hereditarias (hipercolesterolemia familiar) incluyen: deficiencia del receptor de Apo B-LDL, Apo B defectuosa familiar y mutaciones con ganancia de la función de PCSK9.3,12
La hipercolesterolemia familiar heterocigótica tiene prevalencia estimada de 1:200 a 1:500 en la población general.2 Los pacientes deben tratarse con estatinas combinadas con un segundo fármaco (resinas quelantes de colesterol o ezetimiba);13 sin embargo, a pesar de esto, muy pocos alcanzan las metas terapéuticas y se encuentran en alto riesgo cardiovascular.
La forma homocigótica es una enfermedad mucho más rara (1:1,000,000). Los pacientes no responden a medicamentos orales, ya que según el tipo de mutación en el gen del receptor de LDL (R-LDL), algunos son incapaces de producir receptores funcionales, mientras que otros generan receptores con apenas 5 a 25% de su actividad normal.13
De los datos anteriores puede concluirse que para reducir el riesgo cardiovascular residual es imperativo el desarrollo de nuevos fármacos más potentes y seguros, que tengan un espectro de acción farmacológica más amplia y que faciliten el apego de los pacientes al tratamiento.
Biología de la molécula PCSK9
El descubrimiento del papel de la proteína proconvertasa subtilisina-kexina PCSK9 en el metabolismo de C-LDL ha impulsado el desarrollo de nuevas terapias capaces de reducir el colesterol unido a estas lipoproteínas. Los inhibidores de PCSK9 tienen la capacidad de reducir de manera importante las concentraciones de C-LDL, ya sea como monofármacos o combinados con estatinas. Aunque aún no se ha demostrado de manera concluyente, estos medicamentos tienen el potencial de generar un importante beneficio cardiovascular, particularmente en pacientes en riesgo alto.4
El descubrimiento y clonación de la molécula de PCSK9 se logró en el año 2001; posteriormente, Seidah caracterizó las mutaciones con ganancia de la función del gen de PCSK9 relacionadas con mayor riesgo cardiovascular.14 Apenas un año después se identificaron familias afectadas por una mutación con pérdida de la función de este gen que les confería protección cardiovascular. Estos hallazgos detonaron la búsqueda de maniobras farmacológicas para reducir la concentración de PCSK9 y de esta manera emular las mutaciones con pérdida de la función de este gen.14
La PCSK9 es integrante de la familia de enzimas proconvertasas. Las primeras ocho tienen funciones enzimáticas catalíticas, como la activación de precursores hormonales. En contraste, la PCSK9 es la única hasta ahora descrita con una función no catalítica asociada con la regulación del reciclaje del R-LDL.15
Se sintetiza principalmente en el hígado y en menor grado en el intestino. Inicialmente se sintetiza un precursor de 692 aminoácidos: pro-PCSK9, con peso molecular de alrededor de 75 kDa (Figura 2).15,16 El pro-PCSK9 está integrado por los siguientes dominios: 1) péptido señal (aminoácidos 1-30), que permite que sea secretada; 2) prosegmento (aminoácidos 31-152), que funciona como molécula chaperona; 3) dominio catalítico (aminoácidos 153-421), que contiene la tríada catalítica de histidina, serina y aspartato; se exocita y su actividad enzimática se bloquea por el prosegmento cuando es secretada; 4) dominio H (aminoácidos 422-439), que realiza la función de bisagra; y 5) dominio CHR (aminoácidos 440-692), rico en cistidina e histidina.16,17
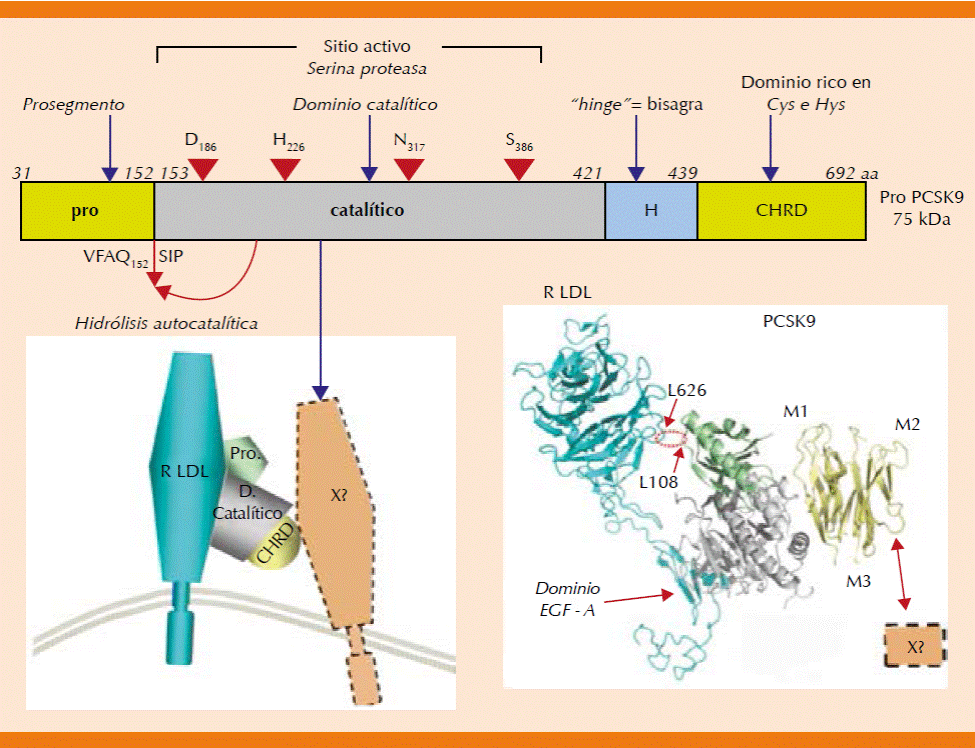
Figura 2 Procesamiento de la PCSK9. Se muestran los distintos dominios de la proteína precursora de la PCSK9 activa (pro-PCSK9). El dominio catalítico es una serina proteasa cuyo único sustrato es la misma proteína. En la figura se observa el sitio exacto de la hidrólisis autocatalítica que separa el prodominio de la cadena proteica principal. El prodominio una vez liberado se une no covalentemente al dominio catalítico bloqueando el sitio activo. La PCSK9 es secretada en esta forma carente de actividad enzimática. La PCSK9 interactúa con el receptor de la apo B100 en el dominio homólogo al factor de crecimiento epidérmico (EGF-A). El conjunto PCSK9-receptor-Apo B-LDL es endocitado en vesículas de clatrina de la superficie celular y destinado a su proteólisis lisosomal. Se muestra un diagrama de la interacción PCSK9-receptor. La estructura tridimensional de estas proteínas sugiere la participación de al menos una proteína transmembrana adicional hasta ahora no identificada (proteína X). Del lado derecho se muestra la interacción de estas proteínas en un modelo de listones tridimiensionales. Tomada de la referencia 14.
Dentro del retículo endoplásmico, el pro-PCSK9 pasa por un procesamiento de hidrólisis autocatalítica, que consiste en que el dominio catalítico hidroliza el enlace peptídico que lo une con el prosegmento o prodominio. El prodominio liberado se une entonces nuevamente, ahora por interacciones débiles, al dominio catalítico y bloquea el sitio activo de la PCSK9. Es decir, el único sustrato conocido hasta ahora de la PCSK9 es ella misma. Este proceso autocatalítico permite la maduración y secreción de la proteína.16
La interacción de la PCSK9 con el receptor de la ApoB100-LDL ocurre por dos vías: 1) intracelular, que consiste en la unión de la PCSK9 al receptor antes de que ambos sean secretados, dirigiéndolo a su destrucción en vesículas intracelulares, y 2) extracelular, la PCSK9 es secretada a la circulación y entonces se une al receptor de LDL ubicado en los huecos recubiertos de clatrina de la superficie celular; posteriormente se endocita el complejo receptor-LDL-PCSK9 y los componentes proteicos de ambos se hidrolizan, lo que evita el reciclaje del receptor a la membrana. Es evidente que la manipulación farmacológica dirigida a reducir la PCSK9 intra o extracelularmente favorece la supervivencia y reciclaje del receptor y, en consecuencia, disminuye las concentraciones de C-LDL (Figura 3).16,17
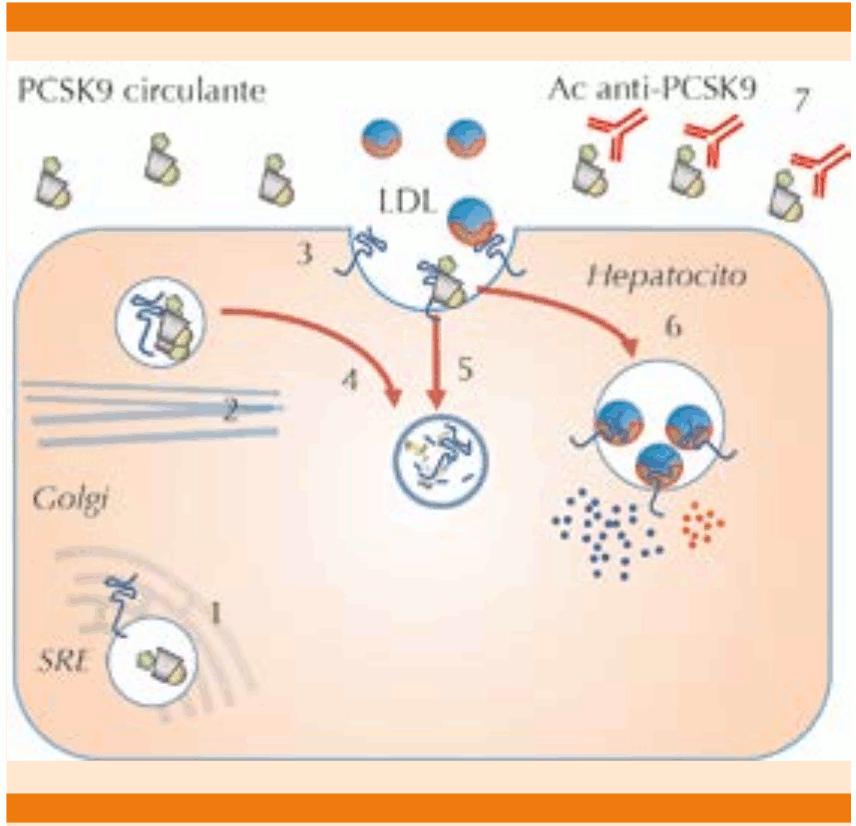
1. La molécula de PCSK9 es sintetizada en el retículo endoplásmico (SRE) junto con el receptor de LDL. 2. Ambas moléculas son llevadas hacia la membrana en una vesícula recubierta de clatrina y son modificadas en el aparato de Golgi. 3. La PCSK9 es liberada a la circulación y el receptor de Apo B-LDL es anclado en la membrana, especialmente en los llamados “huecos recubiertos” de clatrina. 4. La vía intracelular permite que al interactuar la PCSK9 con el receptor de LDL en las vesículas intracelulares, éste sea dirigido a su proteólisis lisosomal antes de ser secretado. 5. La PCSK9 extracelular se une al receptor de LDL, esto modifica al receptor dirigiéndolo también a su destrucción lisosomal evitando su reciclaje. 6. En ausencia de PCSK9 el complejo receptor-LDL es endocitado y mientras que el receptor es reciclado a la membrana, la Apo B es hidrolizada y el colesterol contenido en las LDL es liberado en la célula. 7. Los anticuerpos monoclonales anti-PCSK9 se unen a la proteína circulante impidiendo su interacción con el receptor de Apo B, esto permite la supervivencia del receptor y consecuentemente se reduce la LDL plasmática. Modificada de las referencias 14 y 31.
Figura 3. Mecanismo de acción de los anticuerpos monoclonales anti PSCK9.
La interacción de la PCSK9 con el R-LDL ocurre en el dominio semejante al factor de crecimiento epidérmico (EGFA) de este receptor. Este agregado proteico, al parecer, evita que el R-LDL tome su conformación cerrada definitiva, y al quedar en forma extendida es susceptible de degradación lisosomal.2,16
Cuando el colesterol intracelular disminuye, inicia una secuencia que incluye la activación y translocación del factor de transcripción SREBP2 (proteína de unión al elemento regulador de esteroles). Esta proteína activa al gen SRE, lo que resulta en la síntesis de mayor cantidad de R-LDL y HMGCoA reductasa, la enzima limitante de la biosíntesis endógena del colesterol; sin embargo, simultáneamente, se activa también la síntesis de PCSK9, lo que parece tener un efecto contrarregulador. Como las estatinas bloquean la síntesis de colesterol y de esta manera favorecen la activación de los receptores de LDL, la PCSK9, cuya síntesis se activa simultáneamente, podría limitar sus efectos, ya que ésta orienta al receptor de ApoB-LDL hacia su destrucción.2,16
Las mutaciones con ganancia de función del gen de la PCSK9 dan lugar a una forma de hipercolesterolemia primaria, autosómica dominante, acompañada de concentraciones de C-LDL marcadamente elevadas y enfermedad aterosclerótica prematura. En 2005, Cohen y colaboradores descubrieron la contraparte de este defecto genético al describir mutaciones con pérdida de la función del gen PCSK9 que están relacionadas con concentraciones menores de C-LDL. Estas mutaciones tienen efecto protector contra la enfermedad cardiovascular, que es aún mayor que el que puede lograrse a través de fármacos, debido a que cuando se trata de una causa genética, la concentración de colesterol es baja durante toda la vida, e incluso 40% menor que la que distingue a sujetos sin la mutación.5
En el estudio ARIC17 (de cohortes, longitudinal, birracial) se analizaron distintas variaciones en la secuencia del gen de PCSK9 sobre la incidencia de enfermedad coronaria. Se encontró que 3% de los sujetos de la población abierta de raza negra tenían una de las dos mutaciones sin sentido (PCSK9Y142X y PCSK9C679X) asociadas con pérdida de la función del gen de la PCSK9, que se vincularon con reducción de 28% del C-LDL y de 88% en el riesgo de cardiopatia coronaria.17
En 3% de la cohorte de sujetos de raza blanca se identificó una variación de la secuencia de PCSK9 (PCSKR946L), que se vinculó con disminución de 15% en las concentraciones plasmáticas de C-LDL. A pesar de su efecto más moderado en la reducción del C-LDL, la incidencia de enfermedad coronaria se redujo en 47% (Cuadro 1).17

*p: =0.03; **p: =0.003.
Tomado de las referencias 29 y 30.
Cuadro 1 Mutaciones de pérdida de la función del gen PCSK9 y su efecto en el riesgo cardiovascular
En un estudio de diseño similar efectuado en Dinamarca con 45,699 sujetos, se encontró la mutación PCSKR946L en 3% de ellos, con reducción del C-LDL de 11-16% y del riesgo cardiovascular de 30%.17
Estos datos indican que la disminución moderada del C-LDL a lo largo de la vida (carga vitalicia de colesterol) se asocia con reducción sustancial de la incidencia de eventos coronarios, incluso en poblaciones con prevalencia elevada de factores de riesgo cardiovascular no relacionados con las concentraciones de lípidos (edad, sexo, distribución del índice de masa corporal y prevalencia de hipertensión, diabetes y tabaquismo).17
Anticuerpos anti-PCSK9
En 1975 los investigadores Georges Köhler y César Milstein describieron la técnica del cultivo de hibridomas a partir de la fusión de linfocitos B con células de mieloma múltiple murino y obtuvieron la información necesaria para la síntesis de un anticuerpo específico de manera indefinida; recibieron el premio Nobel de Fisiología y Medicina en 1984.18,19 Después del advenimiento de los anticuerpos monoclonales, inicialmente con secuencias murinas, se desarrolló la tecnología adecuada para la producción de anticuerpos humanos para reducir PCSK9.13 La eliminación de PCSK9 de la circulación aumenta la disponibilidad del R-LDL y, por tanto, disminuye notablemente (50-60%) las concentraciones plasmáticas de C-LDL.1
Los inhibidores de PCSK9 tienen la capacidad de reducir 25 a 30% los valores plasmáticos de Lp(a), esto representa una ventaja con respecto al efecto de las estatinas y sugiere una dependencia, al menos parcial, del catabolismo de Lp(a) a través de R-LDL.1
En el estudio GLAGOV (multicéntrico, doble ciego, con distribución al azar y controlado con placebo para evaluar el efecto del tratamiento con evolocumab) la administración de anticuerpos anti-PCSK9 (evolocumab) en conjunto con las dosis máximas toleradas de estatinas produjo reducción adicional a estatinas del C-LDL de 54 a 75%.4 El beneficio de la importante reducción de las LDL en el proceso aterogénico pudo confirmarse mediante estudios de imagen seriados de las placas de ateroma, que mostraron regresión de éstas en el grupo tratado con evolocumab.
Los anticuerpos humanos monoclonales se generan a través de dos líneas de células madre murinas. A la primera se le inserta el gen que codifica para la inmunoglobulina humana y se conserva el otro alelo de inmunoglobulinas murinas. En la segunda línea se inactivan ambos genes de inmunoglobulina. Cada línea celular da origen a ratones transgénicos (línea híbrida y línea agammaglobulinémica); del cruce de estas líneas, se crea un ratón con la capacidad de producir únicamente IgG humanas xenomouse). A partir del bazo de este ratón, se obtienen linfocitos B para producir hibridomas en cultivo celular. Estos hibridomas permiten generar anticuerpos monoclonales humanos específicos anti-PCSK9.20
La porción variable de los anticuerpos monoclonales se une a PCSK9 para inhibir su función e impedir su interacción con el receptor de LDL. Los anticuerpos anti-PCSK9 se han modificado en su porción Fc para evitar que sea identificada por receptores celulares y active los linfocitos
T, y de este modo no se genere citotoxicidad mediada por células.21
Inhibición de PCSK9 circulante con anticuerpos monoclonales
En 2015, los anticuerpos monoclonales humanos alirocumab (Praluent®, Sanofi) y evolocumab (Repatha®, Amgen) fueron los primeros en ser aprobados para su uso clínico en Europa y Estados Unidos. Se trata de moléculas que se administran por vía subcutánea en régimen quincenal o mensual.1
El evolocumab es un anticuerpo monoclonal totalmente humano, IgG2, con alta afinidad de unión para PCSK9. En estudios de fase II se analizó un rango progresivo de dosis y se encontró que 140 mg cada dos semanas o 420 mg mensuales eran regímenes óptimos para reducir con gran eficiencia las concentraciones de C-LDL.4
En el estudio Descartes se probó la eficacia de evolocumab vs placebo de acuerdo con la terapia de fondo con estatinas. Se observó disminución de C-LDL de entre 50 y 60%, y fue de magnitud similar en todos los grupos, independientemente de las dosis de atorvastatina (10 a 80 mg/día o de la adición de 10 mg de ezetimiba).22
En un estudio de 24 semanas en el que se compararon alirocumab y ezetimiba como monoterapia, los pacientes tratados con alirocumab mostraron reducción de C-LDL de 47 vs 16% de los que habían sido tratados con ezetimiba.16
En un metanálisis de 24 estudios realizados con 10,159 pacientes, se compararon los anticuerpos monoclonales anti-PCSK9 con placebo o ezetimiba. Los pacientes que recibieron anticuerpos anti-PCSK9 mostraron reducciones altamente significativas de C-LDL (47%), colesterol total (39%) y lipoproteína a (26%) e incremento también significativo de C-HDL (6%) al compararlos con el subgrupo que recibió únicamente placebo o ezetimiba.16,23
En el congreso europeo de la Sociedad de Cardiología 2016 celebrado en Roma, Italia,24 se dio a conocer el estudio Odyssey Escape, que reveló que el tratamiento con alirocumab reduce la necesidad de aféresis lipoproteica en pacientes con hipercolesterolemia familiar heterocigótica. Al final del estudio de 18 semanas, en los pacientes tratados con alirocumab descendió 75% la necesidad de aféresis en comparación con los que recibieron placebo.24
El efecto del tratamiento con anticuerpos monoclonales anti-PCSK9 en la morbilidad y mortalidad totales y cardiovasculares aún no se ha determinado de manera concluyente; sin embargo, los resultados observados hasta ahora son favorables. En el metanálisis mencionado se reportó una reducción significativa de la probabilidad de sufrir infarto de miocardio y en la mortalidad total, y una reducción no significativa de la mortalidad cardiovascular en el grupo tratado con anticuerpos vs placebo o ezetimiba.23
Los resultados de los grandes ensayos clínicos de los inhibidores de la PCSK9 determinarán su posible efecto en la morbilidad y mortalidad cardiovasculares, y se darán a conocer en 2018.4,23
Perfil de seguridad de los anticuerpos anti-PCSK9
Al comparar pacientes en tratamiento con anticuerpos anti-PCSK9 con controles en estudios en fases II y III se demostró que existe una incidencia similar de efectos adversos. En ninguno de los ensayos clínicos de fase III se han observado anticuerpos neutralizantes que pudieran disminuir la eficacia de los anticuerpos monoclonales humanos anti-PCSK9.23
Se reportó una prevalencia de aproximadamente 5% de reacciones locales leves en el sitio de inyección de los anticuerpos en los brazos en que se administraba el tratamiento. En pacientes que descontinuaron el tratamiento con estatinas por síntomas musculares, el evolocumab y el alirocumab mostraron seguridad y tolerancia adecuadas.23
Los anticuerpos anti-PCSK9 pueden elevar ligeramente las concentraciones séricas de creatina fosfocinasa (CPK); sin embargo, este incremento es de la misma magnitud que el observado con ezetimiba.23
En estudios iniciales se observó disfunción neurocognitiva leve en pacientes tratados con alirocumab o evolucumab. Sin embargo, en el estudio a largo plazo ODYSSEY (1% de los sujetos que recibieron anticuerpos vs 0.5% de los que tomaron placebo; p=0.17), al ampliar el nú mero de pacientes se consideró que este efecto es infrecuente y clínicamente poco significativo.24
En octubre de 2016, de manera sorpresiva, la casa farmacéutica Pfizer anunció que suspendía el desarrollo de bococizumab, un anticuerpo humanizado anti-PCSK9, debido a pérdida de eficacia a largo plazo, posiblemente por inmunogenicidad, ya que se trataba de un anticuerpo con secuencias murinas. Asimismo, el laboratorio argumentó varias razones de mercado para suspender el desarrollo de este producto.25
Quizá la mayor desventaja de los anticuerpos anti-PCSK9 hoy día es su elevado costo, espe cialmente en países en desarrollo en los que pocos pacientes tienen cobertura completa de servicios médicos.
Nuevas estrategias para disminuir la PCSK9
Hace poco se describió la posibilidad de inhibir la síntesis de PCSK9 al administrar moléculas de ácido ribonucleico (ARNsi) de bajo peso molecular de "silenciamiento" (ARNsi). El ARNsi se acopla a la vía natural del ARN de interferencia (ARNi) al unirse al complejo silenciador inducido por ARN (RISC). De este modo, el ARN mensajero (ARNm) que codifica para la PCSK9 es escindidio, con lo que se inhibe la traducción de la PCSK9. A diferencia de los anticuerpos anti-PCSK9 que sólo interfieren con las acciones extracelulares de la proteína, el mecanismo de acción de los ARNsi permite la inhibición de las acciones intra y extracelulares de la PCSK9. Esto evitaría la regulación a la alza de la síntesis de PCSK9 inducida por estatinas.26
El ensayo clínico ORION-126 es un estudio controlado, doble ciego y con distribución al azar, realizado con voluntarios sanos en el que se analizó el inclisirán, un ARNsi unido a una nanopartícula lipídica (ALN-PCSsc), que puede administrarse por vía subcutánea y con acción prolongada. Interfiere con la traducción del ARNm de la PCSK9 y es conjugado con carbohidratos de N-acetilgalactosamina que permiten que sea capturado por los abundantes receptores de asialoglucoproteínas abundantes en el hígado. El estudio mostró que una dosis única de inclisirán de 300 mg por vía subcutánea redujo la concentración de C-LDL más de 50% por periodos de cuatro a seis meses. El inclisirán fue bien tolerado y la incidencia de reacción en el sitio de inyección resultó menor de 5%. Los efectos adversos más comunes fueron: tos, dolor musculoesquelético, nasofaringitis, cefalea y diarrea; no se observaron efectos adversos graves. Aunque el estudio se realizó con un número reducido de participantes sanos, los resultados son alentadores y al parecer la relación riesgo-beneficio es aceptable.26
Hace poco se publicaron los resultados de un estudio de "aleatorización mendeliana"27 como una forma de probar que la reducción de origen genético de la PCSK9 y la consiguiente disminución del C-LDL tiene un efecto favorable en el riesgo cardiovascular. El estudio mostró que la acción de variantes genéticas en el gen de la PCSK9 y de la enzima hidroximetilglutaril CoA reductasa (HMGCoAR), que regula la síntesis endógena del colesterol en el riesgo de sufrir enfermedad cardiovascular se atribuye a los efectos que tienen en la concentración de C-LDL y no a través de algún otro efecto de tipo pleio-trópico.27 Los datos del estudio son importantes como "prueba de concepto" que justificaría los protocolos que actualmente están en proceso para reducir la PCSK9 por medios farmacológicos a largo plazo.
Inmunización activa anti-PCSK9
La interesante posibilidad de inducir en el sistema inmunológico del paciente una respuesta de rechazo contra una proteína propia como la PCSK9 representa un reto formidable.
El proyecto consiste en desarrollar una verdadera vacuna (inmunización activa) capaz de activar a las células B y hacerlas autorreactivas hacia esa proteína, en este caso la PCSK9.
Esto puede lograrse mediante la alteración de la proteína propia con epítopes antigénicos, para que al exponerse a las células B, éstas se vuelvan autorreactivas y a largo plazo se produzcan células plasmáticas de memoria capaces de generar anticuerpos específicos contra la proteína deseada.
Una vacuna desarrollada por el grupo Affiris, en Viena, utiliza un epítope foráneo que es la hemocianina, que se adhiere a segmentos de la PCSK9. En experimentos con primates no humanos se ha logrado la producción endógena de anticuerpos anti-PCSK9 que persisten incluso por un año.28
Los epítopes más utilizados hasta ahora son partículas de un virus inactivado que contiene segmentos de PCSK9. El complejo se administra como una vacuna convencional para activar la respuesta del sistema inmunológico y producir endógenamente anticuerpos anti-PCSK9.28
Estudios recientes realizados en animales muestran que estas vacunas pueden inducir una respuesta inmunológica de alta afinidad y especificidad de anticuerpos anti-PCSK9 en todas las especies analizadas hasta la fecha. La tolerancia y efectividad de la vacuna para reducir la PCSK9 y el C-LDL se ha probado en primates no humanos con resultados satisfactorios y persistentes por periodos prolongados (seis meses).28
CONCLUSIONES
Apenas 14 años transcurrieron desde la clonación de la PCSK9 en 2001, hasta la aprobación en 2015 para uso terapéutico de los anticuerpos monoclonales evolocumab y alirocumab dirigidos a bloquear la PCSK9 circulante, para el tratamiento de los pacientes con hipercolesterolemias primarias o que no alcanzaron las metas terapéuticas con estatinas.
Los anticuerpos monoclonales anti-PCSK9 son medicamentos eficaces para lograr importantes reducciones del C-LDL (del orden de 50%). Estos resultados se obtienen después de indicar las dosis máximas toleradas de estatinas, especialmente en pacientes en riesgo cardiovascular alto. Los efectos colaterales hasta ahora han sido mínimos y aceptables, dado el gran beneficio de reducir en forma tan considerable el C-LDL, e incluso la lipoproteína a.
Aunque los datos preliminares parecen confirmar la esperada reducción de la morbilidad y mortalidad cardiovasculares, aún no se han realizado estudios del tamaño suficiente para demostrar el beneficio cardiovascular a largo plazo que se espera se derive de estos medicamentos. En la actualidad, la limitación más importante para difundir el tratamiento con anticuerpos anti-PCSK9 es su elevado costo económico.
La inhibición de PCSK9 a través de moléculas diferentes a los anticuerpos monoclonales, como con el uso de ARNsi dirigidos a interferir con la traducción del ARN mensajero del gen de la PCSK9, podría ser en el futuro una alternativa interesante para bloquear aún más los efectos intracelulares de esta proteína y permitir mayor supervivencia de los receptores de LDL y, en consecuencia, reducir C-LDL con una posología más simple (administración trimestral o semestral).
La historia de la PCSK9 es un ejemplo fascinante de la rapidez con la que las ciencias básicas permiten el desarrollo y utilización terapéutica de fármacos para abatir grandes problemas de salud pública en el mundo.

