











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La tirosinemia tipo 1 o tirosinemia hepatorrenal (TYR-1) (OMIM 276700) es un error innato del metabolismo de los aminoácidos ocasionado por deficiencia de la fumarilacetoacetato hidrolasa, última enzima del ciclo catabólico de la tirosina (Figura 1). Su prevalencia global es de uno en 100,000 a 120,000 recién nacidos vivos,1,2 aunque existen áreas geográficas como Quebec, Canadá, donde la frecuencia es mucho mayor, uno en 1846 recién nacidos vivos.3,4 En México, la prevalencia se desconoce por la falta de tamiz neonatal obligatorio para tirosinemia hepatorrenal, por lo que no existe un registro nacional de casos.
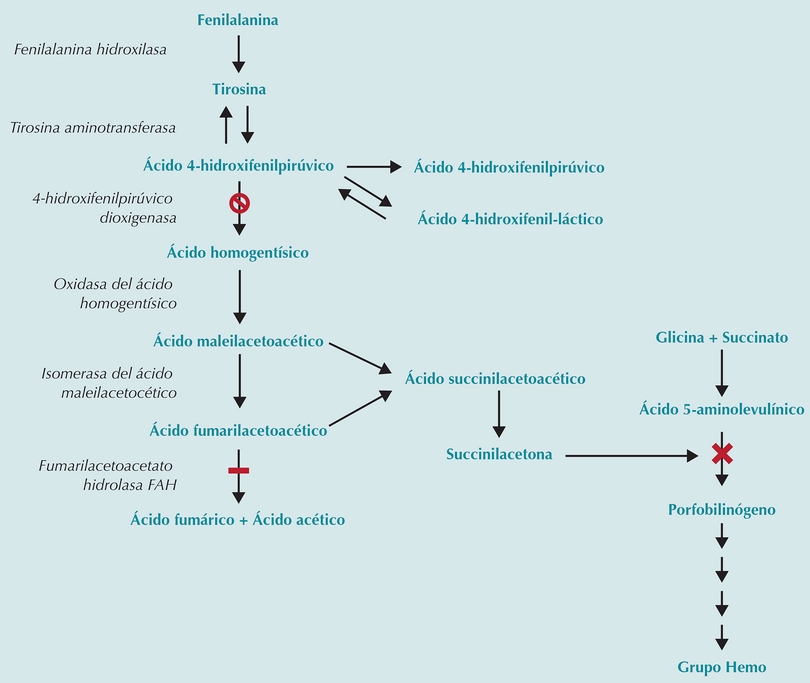
Figura 1 Ruta metabólica de la tirosina. La barra negra señala el sitio del bloqueo de la enzima fumarilacetoacetato hidrolasa, responsable de la tirosinemia hepatorrenal; la cruz muestra el sitio de inhibición del metabolismo del grupo Hemo por acumulación de succinilacetona. El símbolo ⍉ señala el sitio de acción de la nitisinona.
El cuadro clínico se caracteriza por falla hepática con coagulopatía pronunciada acompañada de ascitis y edema, así como signos de raquitismo hipofosfatémico secundarios a tubulopatía renal con síndrome de Fanconi. También existe transaminasemia e hipoalbuminemia y los pacientes pueden o no tener ictericia, glucosuria, aminoaciduria y fosfaturia.4 A mediano y largo plazos, pueden ocurrir nefrocalcinosis, gloméruloesclerosis, falla renal crónica, afección de nervios periféricos, cirrosis, nódulos hepáticos y carcinoma hepatocelular.5,6 La tirosinemia hepatorrenal se clasifica según la edad de presentación de síntomas: aguda, cuando los síntomas se presentan en los seis primeros meses de vida; subaguda, que se inicia entre los seis y los 12 meses de edad y crónica, después del año de vida. Mientras más temprano comienzan los síntomas, más severa es la enfermedad.
Es bien sabido que cuando estos pacientes no se diagnostican en el periodo neonatal y no reciben tratamiento temprano, tienen una alta mortalidad.7-9 En México se ha descrito la elevada mortalidad de la tirosinemia hepatorrenal.10 La succinilacetona es el metabolito patognomónico de la tirosinemia hepatorrenal11 y el patrón bioquímico de la enfermedad incluye hipertirosinemia, hipermetioninemia, acumulación de los ácidos fumarilacetoacético y maleilacetoacético en la sangre y en orina, los cuales juegan un papel crucial en el daño hepático, renal y neurológico de los pacientes. Como indicador, la elevación aislada de tirosina carece de sensibilidad, ya que puede deberse a un gran número de patologías o condiciones médicas como el ayuno, uso de nutrición parenteral total, tirosinemia transitoria del neonato y hepatopatías en general, de causa congénita o adquirida.1,9 Es muy importante considerar que la alfa-fetoproteína, que es un biomarcador tumoral, suele tener niveles promedio de 160,000 ng/mL en etapas muy tempranas del padecimento.1,4-8
Una de las complicaciones más graves de la tirosinemia hepatorrenal es la presencia de crisis neurológicas o pseudoporfiria (“porphyria-like”). Aunque algunos autores las han descrito desde hace cerca de 50 años,12,13 la primera descripción clínica y bioquímica detallada fue realizada por Mitchell y sus colaboradores en 1990.14 La sintomatología principal de estas crisis es el dolor en las piernas o abdominal, usualmente acompañado de vómito e íleo paralítico. Existe hipertonía típicamente axial, que varía en severidad, desde una leve resistencia a la flexión del cuello hasta una postura de opistótonos. También ocurre una neuropatía motora aguda ascendente, que puede progresar a parálisis con dificultad respiratoria que requiere ventilación mecánica.6 En algunos casos ocurren episodios de automutilación caracterizadas por mordeduras y laceración de la lengua o bruxismo severo que causa avulsión de los dientes. También hay hipertensión arterial y taquicardia.
Los principales datos de laboratorio durante las crisis neurológicas de la tirosinemia hepatorrenal son hiponatremia, hipokalemia, aumento de enzimas hepáticas, tiempo de protrombina prolongado y niveles elevados de ácido delta-aminolevulínico. Ocasionalmente hay hiperamonemia y bicarbonato sérico bajo.14
Bioquímicamente, las crisis neurológicas en esta enfermedad se deben a que la acumulación de succinilacetona inhibe la síntesis de porfirina a nivel de la enzima deshidratasa del ácido aminolevulínico y causa acumulación de ácido 5-aminolevulínico (Figura 1), que es un potente neurotóxico responsable de la sintomatología similar a la de la porfiria aguda.1
La nitisinona o NTBC (2-(2-nitro-4-trifluorometilbenzoil)-1,3-ciclohexanediona) (Orfadin®) es un potente inhibidor de la enzima 4-hidroxifenilpiruvato dioxigenasa (4-HPPD), evitando la degradación de la tirosina y la producción de succinilacetona (Figura 1); desde 1992 se convirtió en el tratamiento de elección de la tirosinemia hepatorrenal, utilizada de manera conjunta con la dieta restringida en tirosina. Esta combinación ha reducido la necesidad de trasplante hepático o hepatorrenal y ha mejorado notablemente las tasas de supervivencia de los pacientes.11,15,16 Se sabe que tan sólo 24 horas después de la primera dosis de nitisinona la succinilacetona no debe ser detectada en la orina, ni en la sangre; debe encontrarse por debajo del valor de referencia; la coagulopatía suele revertir en 48 horas y los tiempos de coagulación se normalizan en una semana. La disminución de alfa-fetoproteína suele ser logarítmica y tiende a normalizarse después del primer año de tratamiento continuo.4 La dosis de nitisinona debe ser individualizada según el peso y el cuadro clínico.
El tratamiento de la tirosinemia hepatorrenal requiere de un plan de nutrición individualizado con modificaciones de acuerdo a la ganancia pondo-estatural del paciente, conforme a las concentraciones de aminoácidos en sangre, particularmente las de tirosina y fenilalanina, siguiendo las recomendaciones de Acosta y sus colaboradores.17 Las evaluaciones nutricias se realizan con el parámetro de talla para la edad, y dado que el peso en los pacientes con tirosinemia hepatorrenal puede estar afectado por el edema. La hepatomegalia y la ascitis, el perímetro del brazo y el pliegue cutáneo tricipital se toman como marcadores de repleción nutricional.
CASO CLÍNICO
Niña de cuatro años de edad, producto de un embarazo normoevolutivo no planeado, de padres no consanguíneos aparentemente sanos. Tuvo el antecedente de dolor abdominal e íleo a los ocho meses de edad, que requirió hospitalización por ocho días, manejado como probable oclusión intestinal y que se resolvió con ayuno. A la edad de 9 meses los padres notaron aumento del perímetro abdominal y estreñimiento, por lo que acudieron a un hospital general en donde diagnosticaron intolerancia a la lactosa, que fue tratada con laxantes y fórmula libre de lactosa, sin respuesta favorable. Un ultrasonido abdominal reveló hepatomegalia, por lo cual fue enviada a un servicio de pediatría donde se hallaron pruebas de función hepática anormales, plaquetopenia y tiempos de coagulación prolongados que requirieron transfusión de tres unidades de plasma, con el diagnóstico de hepatitis de etiología a determinar. Una biopsia hepática fue sugestiva de tirosinemia hepatorrenal; sin embargo, no recibió tratamiento específico.
A los dos años de vida fue enviada a nuestra institución, donde se corroboró el diagnóstico de tirosinemia hepatorrenal por la presencia de succinilacetona de 3.6 µM (V.R. < 1.0 µM), tirosina 342 µM (V.R. < 169 µM) en sangre, así como alfafetoproteína de 45,364 UI/mL (V.R. 0.5-5.5 UI/mL). Se le trató con dieta restringida en tirosina y alimento libre de fenilalanina y tirosina (Tyrex-2) (Cuadro 1), así como nitisinona: 1 mg/kg/día, que inicialmente recibió por 11 meses. Sin embargo, como dicho medicamento pertenece a la categoría de fármacos huérfanos, su adquisición por nuestra institución fue compleja y la paciente se quedó sin tratamiento por un mes.
Cuadro 1 Planes de alimentación utilizados en el presente caso A): inicial, B): seguimiento
| A) Dieta inicial para resolver la emergencia, de 720 kcal, con una osmolaridad de 685 mOsm/L, para la paciente de 3 años 11 meses, con un peso de 17.6 kg y talla de 100.8 cm | |||||||
| Alimento | Cantidad | PHE/mg | TYR/mg | Proteína/g | Energía/kcal | ||
| Fórmula sin fenilalanina ni tirosina | 150 g | -- | -- | 22.5 | 720 | ||
| Agua | 600 mL (20 oz) | -- | -- | -- | |||
| Total | -- | 22.5 | 720 | ||||
| Recomendaciones 1-4 años17 | 450-1050 | >30 | 900-1800 | ||||
| B) Dieta de seguimiento utilizada, de 1700 kcal, con una osmolaridad de 732 mOsm/L, para la paciente con 4 años cumplidos, relación calórico proteica de 47:1; 101 kcal/kg/d; proteína: 2.1 g/kg/d | |||||||
| Alimento | Cantidad | PHE/mg | TYR/mg | Proteína/g | Energía/kcal | ||
| Fórmula sin fenilalanina ni tirosina | 160 g | -- | -- | 24 | 768 | ||
| Fórmula de inicio | 21.5 g (5 medidas) | 169 | 2.04 | 112.45 | |||
| Maicena | 30 g | -- | -- | 105 | |||
| Agua | 700 mL ( 23.3 oz ) | -- | -- | -- | |||
| Verdura | 5 Eq | 125 | 2.5 | 50 | |||
| Fruta | 5 Eq | 125 | 2.5 | 300 | |||
| Cereal | 8 Eq | 400 | 4.8 | 240 | |||
| Grasas | 1 Eq | 9 | 0.1 | 60 | |||
| Alimentos libres A17 | 1 Eq | 9 | 0.1 | 65 | |||
| Total | 837 | 36.04 | 1700.45 | ||||
| Recomendaciones 1-4 años17 | 450-1050 | >30 | 900-1800 | ||||
Al principio de la quinta semana de haber suspendido la nitisinona acudió al servicio de urgencias del Instituto Nacional de Pediatría por vómito postprandial y diarrea de dos días acompañada de fiebre de 38ºC en una sola ocasión; dolor abdominal, astenia, adinamia, insomnio e irritabilidad, tos productiva con rinorrea hialina y tiros intercostales. En la exploración física presentó taquipnea, e hipertensión (142/72 mmHg), hepatomegalia de 2 cm por debajo del reborde costal. La hipertensión fue persistente y llegó hasta 130/90 y 142/72 mmHg, lo que requirió tratamiento con prazosina 250 mcg cada 6 h, enalapril, 5 mg cada 24 h, furosemide, 10 mg cada 12 h. No hubo respuesta, por lo que se agregó propranolol 10 mg cada 8 h, losartán 25 mg cada 12 h y amlodipino 5 mg cada 24 h, así como dieta libre en tirosina y fenilalanina.
Los principales resultados de laboratorio se muestran en el Cuadro 2, destacan la hiponatremia y la hipokalemia, así como leve aumento de las enzimas hepáticas, del amonio y de los tiempos de coagulación. El vómito fue persistente y el dolor abdominal requirió opioide (buprenorfina a dosis baja en infusión 4 mcg/kg/día) y clonixinato de lisina 1 mg/kg/dosis IV cada 8 h. Un ecocardiograma mostró hipertrofia ventricular izquierda con función ventricular normal. A los 17 días de internamiento se logró conseguir la nitisinona, que se reinició a 1 mg/kg/día y a las 24 horas después, hubo una notable mejoría clínica, con normalización gradual de la tensión arterial, de los electrolitos séricos y de la succinilacetona (Figura 2). Egresó una semana después del reinicio del medicamento, clínicamente estable.
Cuadro 2 Principales resultados de laboratorio observados al momento del ingreso
| Sodio | 117 | 138-145 mmol/L |
| Potasio | 2.4 | 3.7-5.0 mmol/L |
| Cloro | 107 | 96-109 mmol/L |
| Aspartato amino transferasa | 47 | 20-60 UI/L |
| Alanina amino transaminasa | 58 | 5-45 UI/L |
| Gama-glutamiltransferasa | 41 | 6-19 UI/L |
| Nitrógeno ureico | 11.2 | 5-17 mg/dL |
| Creatinina | 0.47 | 0.2-0.7 mg/dL |
| Glucosa | 63 | 57-115 mg/dL |
| Alfa fetoproteína | 478 | 0.5-5.5 UI/mL |
| Amonio | 50 | 11-35 mol/L |
| Tiempo de protrombina | 13.6 | 10.43–13.63 segundos |
| Porcentaje de tiempo de protrombina | 74 | 76.04–124.02% |

DISCUSIÓN
Antes del advenimiento de la nitisinona las crisis neurológicas de pseudoporfiria de la tirosinemia hepatorrenal ocurrían en 42% de los pacientes y tenían una elevada mortalidad.14 En los pacientes que reciben nitisinona de forma continua se evitan dichas crisis.18,19 En la literatura existen dos trabajos recientes que señalan la presencia de crisis de pseudoporfiria por suspensión de nitisinona, una en un paciente africano de 8 meses de edad y otro turco, ambos habían sido oportunamente diagnosticados con tirosinemia hepatorrenal y tratados desde el periodo neonatal. En estos casos, la interrupción del medicamento se debió a negligencia de los padres.20,21 Llama la atención que, tanto en nuestro caso como en los descritos en la literatura, los síntomas neurológicos se iniciaron entre la 4ª y 5ª semanas después de la interrupción del medicamento; se caracterizaron por dolor abdominal, astenia, adinamia, alteraciones del estado de conciencia, hipertensión arterial acompañados de hiponatremia, hipokalemia, aumento de las enzimas hepáticas y de la succinilacetona. En nuestro paciente, la suspensión del medicamento se debió a la dificultad para adquirir fármacos de alto costo catalogados como huérfanos. En nuestra institución, gracias a la estrategia del Seguro Popular,22 la adquisición de este fármaco ya se ha solucionado.
En concordancia con lo señalado por otros autores,20,21 la resolución de la crisis con la reintroducción de la nitisinona es rápida; los pacientes recobran parámetros bioquímicos (Figura 2) y clínicos normales en menos de una semana. Es importante mencionar que durante el tiempo que esta paciente suspendió el medicamento, mantuvo la dieta libre de tirosina y fenilalanina; sin embargo, se sabe que esto no es suficiente para reducir los niveles de succinilacetona, puesto que deja de ocurrir el efecto inhibidor de la nitisinona sobre la 4-hidroxifenilpirúvico dioxigenasa.17 Otro aspecto a considerar con la suspensión de nitisinona es el aumento del riesgo de desarrollar carcinoma hepatocelular, que es una de las complicaciones más graves de la tirosinemia hepatorrenal.
Finalmente, aunque en nuestro caso el diagnóstico de tirosinemia hepatorrenal ya estaba establecido antes de que aparecieran las crisis tipo porfiria, es importante destacar que en todo paciente pediátrico con dolor abdominal y manifestaciones neurológicas debe hacerse diagnóstico diferencial con otras patologías tales como síndrome de Guillain-Barré, afección por virus neurotrópicos (polio, coxackie, ecovirus, enterovirus, entre otros), así como neurotoxicidad por plomo, arsénico o talio.
CONCLUSIONES
El tratamiento con nitisinona debe ser continuo y no debe suspenderse nunca de manera abrupta, pues la suspensión causa elevación de la succinilacetona y otros metabolitos tóxicos, lo que aumenta el riesgo de padecer crisis neurológicas de pseudoporfiria, las cuales tienen elevada mortalidad; dichas crisis revierten en general una semana después de reiniciar el medicamento.

