











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkCASO CLÍNICO
Niña de 11 meses de edad, originaria de Ecatepec, Estado México. Producto del primer embarazo de madre de 27 años. Nació por cesárea a las 41 semanas con Apgar 8 y 9 al minuto y 5, respectivamente; peso de 3,450 kg con talla de 50 cm. El tamiz neonatal fue normal y el periodo perinatal se reportó sin complicaciones. Se alimentó con fórmula láctea desde el nacimiento, ablactación a los 6 meses con desarrollo neurológico adecuado para su edad. Esquema de vacunación completo. Antecedentes familiares de abuela paterna finada por cáncer pulmonar, el abuelo paterno tuvo cáncer en miembro pélvico no especificado y una tía paterna tuvo cáncer de sistema nervioso central. Actualmente camina con ayuda.
Inició su padecimiento a los 6 meses cuando la madre notó voz ronca, crecimiento de vello púbico, aumento del tamaño de clítoris y edema de pies. Acudió a facultativo quien descartó patología. A los 10 meses de vida presentó pico febril y un pediatra diagnosticó probable hiperplasia suprarrenal; acudió a centro médico en el Estado de México de donde la refirieron al Instituto Nacional de Pediatría (INP) con diagnóstico de tumor virilizante de glándula suprarrenal el 24 de enero del 2014.
A la exploración física su peso fue 9 kg (25-50); talla 71 cm (25); PC 46 cm (75-90); FC 124; FR 26; TA 90/60 y T 36°. La edad aparente es igual a la cronológica, alerta, activa, reactiva, cooperadora, adecuada coloración y buen estado de hidratación. Normocéfala, fontanela anterior puntiforme, ojos simétricos, pupilas normorreflécticas, narinas permeables, cavidad oral bien hidratada. Filtrum prominente con vello delgado. Cuello sin alteraciones. Tórax simétrico, campos pulmonares bien ventilados. Ruidos cardiacos rítmicos y de buen tono e intensidad sin soplos o fenómenos agregados. Abdomen globoso, blando, depresible sin puntos dolorosos, peristalsis presente: se palpó masa en mesogastrio e hipocondrio izquierdo, firme, móvil con superficie regular. Genitales con vello en labios mayores liso y delgado, Tanner III, clítoris aumentado de tamaño con crecimiento de cuerpos cavernosos, labios mayores sin hiperpigmentación, región perianal sin alteraciones. Extremidades íntegras, sin edema, adecuado llenado capilar. Glasgow 15/15 sin datos de focalización.
Laboratorios fuera del Instituto Nacional de Pediatría (22.01.14): Hb 13, Hto 37.8, leucos 8430, N 19%, L 72%, plaq 275,000, Na 138, K 5.3, CL 105, Ca 9.4, P 5.8. Mg 1.8, TP 11.7/100%, TTP 26, gluc 92, CR 0.1, BUN 12, colesterol 111, TG 79, bil total 0.8, bil directa 0.1, bil indirecta 0.7. PT tot 6.2, alb 3.7, globulina 2.5, AST 48, ALT 30, fosf alc 456, AU 3.9, DHL 365, Ag carcinoembrionario 1.23, alfa feto proteína 27.41, beta HGC 0.12, FSH 0.70, LH 0.61, estradiol II: 15, testosterona 4.14, progesterona 2.08, prolactina 11.24, cortisol 10.92.
Resonancia magnética: tumor suprarrenal izquierdo que desplaza riñón izquierdo, de 7 × 5 cm, sin datos a enfermedad a otro nivel. El servicio de Cirugía Oncológica programó resección quirúrgica. El servicio de endocrinología recomendó administración de esteroide a dosis de estrés, por el riesgo de crisis adrenal antes y durante la cirugía. El servicio de genética comentó sobre la posibilidad de un síndrome de Li Fraumeni.
A los tres días de ingreso se realizó adrenalectomía total izquierda. Se identificó tumor suprarrenal izquierdo de coloración blanco amarillento de aproximadamente 7 × 5 cm de aspecto friable pero bien encapsulado. Se logró extraer en su totalidad de la cavidad. Se administró esteroide a dosis de estrés y no se reportaron complicaciones. La evolución post operatoria fue adecuada (Cuadro 1).

HALLAZGOS PATOLÓGICOS
El tumor pesó 30 gramos y midió 5 × 4 × 4 cm. En la cápsula se identificaron vasos delgados visibles (Figura 1). Al corte, el tumor se apreciaba multinodular de consistencia blanda y color blanco grisáceo con áreas amarilla naranja (Figura 2). Microscópicamente, el tumor estaba constituido por células grandes con acentuado pleomorfismo y abundante citoplasma eosinófilo (Figura 3). La neoplasia comprime la corteza suprarrenal por debajo de la cápsula (Figura 4). A mayor aumento es más evidente el pleomorfismo y desde el punto de vista arquitectural el patrón es difuso (Figura 5). Hay mitosis atípicas (Figura 6) y se encuentran más de 5 mitosis por 50 campos de gran aumento. La necrosis presente como áreas eosinófilas en patrón geográfico se identifican en varios de los campos microscópicos (Figura 7). La permeación vascular es uno de los principales criterios de malignidad en los carcinomas de corteza suprarrenal; en la siguiente imagen se ilustra un vaso con un gran agregado tumoral en su luz (Figura 8). Por inmunohistoquímica se encontró un importante índice de proliferación celular, con un Ki67 de 20% que se expresa como positividad nuclear (Figura 9). La proteína p53 igualmente se expresa en forma nuclear en una proporción del 30% (Figura 10). Los criterios histopatológicos de Weiss para caracterizar neoplasias adrenocorticales malignas se ilustran en el Cuadro 2. El tumor que presentamos reúne 7 de los 9 criterios (*).
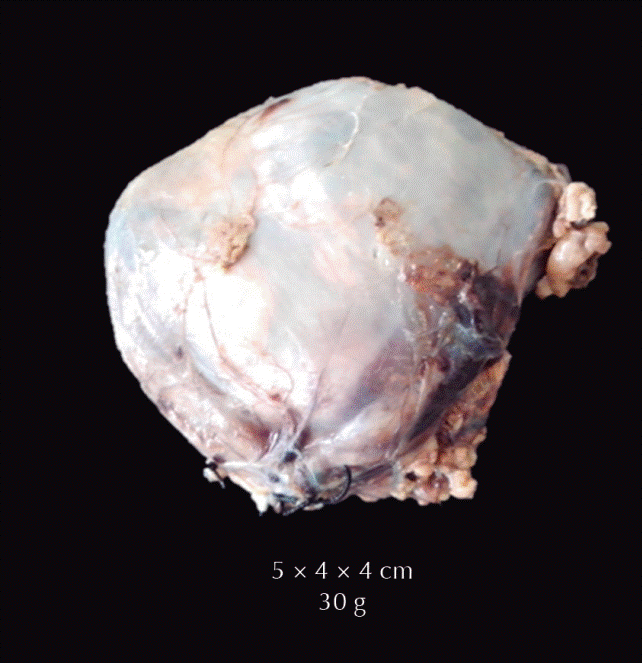
Figura 1 Tumor de 30 gramos de peso y 5 × 4 × 4 cm. En la cápsula se identificaron vasos delgados visibles.
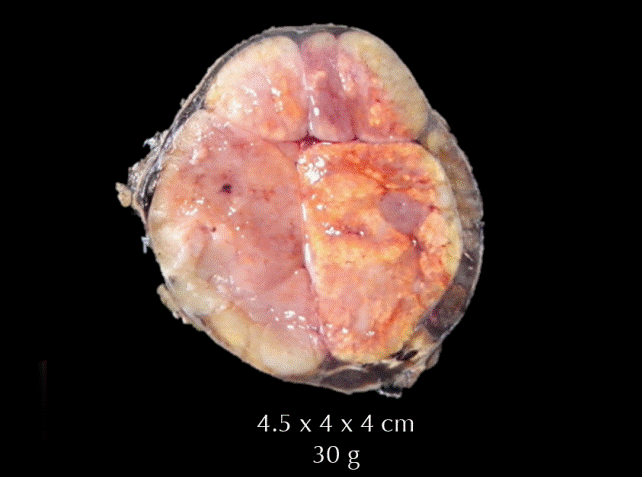
Figura 2 Corte del tumor multinodular: consistencia blanda y color blanco grisáceo con áreas amarilla naranja.
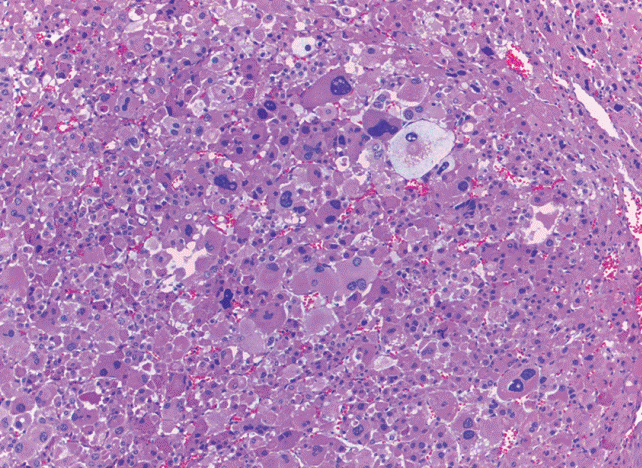
Figura 3 Microscópicamente se observan células grandes con acentuado pleomorfismo y abundante citoplasma eosinófilo.
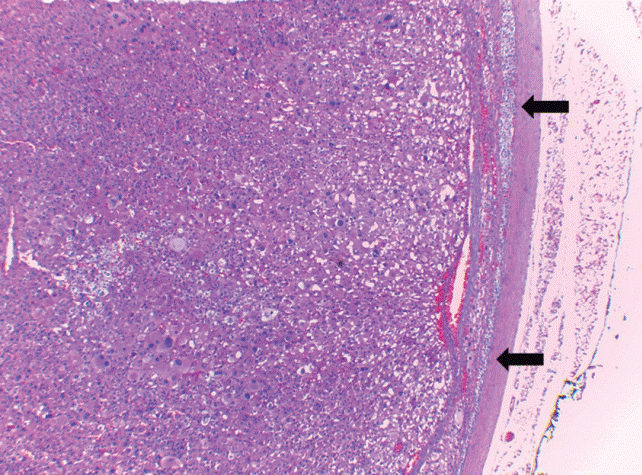
Figura 4 Las flechas señalan a la neoplasia comprimiendo la corteza suprarrenal por debajo de la cápsula.


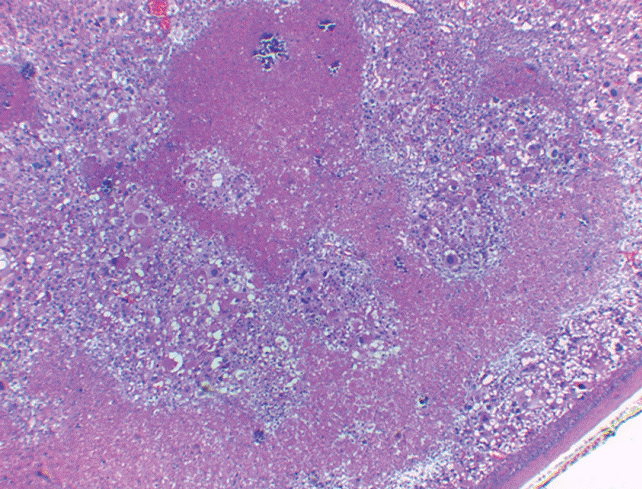

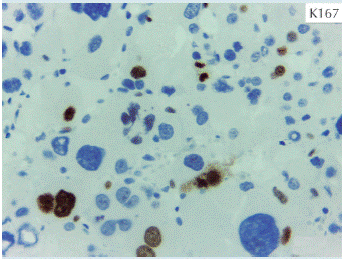
Figura 9 Importante índice de proliferación celular con Ki67 de 20% que se expresa como positividad nuclear.


COMENTARIO
Nuestra paciente inició su patología a los 6 meses de edad con vello púbico, voz ronca y clitoromegalia de aparición brusca. A la exploración física se detectó masa en hipocondrio izquierdo. La primera entidad obligatoria e impostergable a descartar en esta paciente fue un carcinoma suprarrenal, ya que es la patología más común en niños menores de 5 años de edad. En la gran mayoría de los casos (90%) estos tumores son hormonalmente activos, por lo que frecuentemente se manifiestan con virilización aislada secundaria a la excesiva producción de andrógenos suprarrenales, que producen clitoromegalia en las niñas o incremento en el tamaño del pene en los niños; además de pubarca, sudor apócrino, aceleración en el crecimiento y voz ronca.
En algunos casos se encuentra también elevada la producción de cortisol, por lo que los pacientes afectados presentan además síndrome de Cushing con hipertensión arterial, obesidad, facies de luna llena, estrías de color rojo-vinosas, acné y retraso en el crecimiento. La presencia de hipercortisolismo sin hiperandrogenismo es muy rara en este tipo de carcinomas (<5%). La elevación del marcador tumoral alfa fetoproteína refuerza la posibilidad de neoplasia maligna.
Se excluye la posibilidad de hiperplasia suprarrenal congénita, ya que al nacimiento no había clitoromegalia, además de que en esta entidad no se presenta vello púbico en los primeros años. Asimismo, se descartó pubertad precoz por datos clínicos, ya que en una niña el primer dato de pubertad sería el crecimiento de glándula mamaria, consecuencia de la activación ovárica por producción de estrogénos, y no por pubarca que es dependiente de andrógenos suprarrenales y de ningún modo por la presencia de clitoromegalia.
El perfil hormonal prequirúrgico confirmó hiperandrogenismo con niveles muy elevados tanto de testosterona como de sus precursores: 17 hidroxi-progesterona, androstenediona y deshidroepiandrosterona (DHEA). Estas hormonas actúan en los cuerpos cavernosos del clítoris y son responsables de la clitoromegalia y de los otros datos de virilización de la paciente.
El nivel normal de cortisol descartó hipercortisolismo, lo cual era esperado, ya que no tenía datos clínicos del mismo. El reporte de hormona adrenocorticotropa (ACTH) indetectable, probablemente sea secundario a una muestra mal conservada, ya que se debe de tomar con anticoagulante ácido etilendiaminotetraacético (EDTA), centrifugarla en frío y procesarla de inmediato, pues esta hormona es muy sensible a la temperatura y tiene una vida media menor de 10 minutos. El tratamiento con cortisol prequirúrgico no está indicado en los casos que no se documenta hipercortisolismo.
Los estudios de imagen confirmaron el diagnósticodeunamasade7×5cmaniveldeglándula suprarrenal izquierda, por lo que la palpación de masa en hipocondrio izquierdo, era el riñón que se encontraba desplazado por la tumoración suprarrenal.
El diagnóstico diferencial con un adenoma suprarrenal productor de andrógenos es histopatológico, al excluir la invasión a la cápsula adrenal o permeación de los vasos sanguíneos. La tumoración adrenal debe extirparse quirúrgicamente en su totalidad para el diagnóstico definitivo. No existe correlación inequívoca entre el tamaño del tumor o sus características histopatológicas y su capacidad de producción hormonal. La conducta inmediata es la resección tumoral total ya que es el único tratamiento potencialmente curativo, debido a que el carcinoma suprarrenal no es sensible a quimioterapia o radioterapia. Al momento del diagnóstico 60% de los pacientes tienen enfermedad limitada y el tumor puede ser completamente resecado.
En esta paciente la tumoración fue resecada en su totalidad y el estudio histopatológico confirmó carcinoma de la corteza suprarrenal con permeación vascular.
La incidencia de los tumores adrenocorticales en niños es extremadamente baja, con sólo el 0.2% de los tumores malignos en etapa pediátrica durante los primeros 5 años de vida (media de 3-4 años de edad) predominando en el sexo femenino. Es importante detectar esta patología en etapas tempranas ya que la supervivencia a cinco años es de 82% cuando están en etapa I; 61% en etapa II; 50% en etapa III y sólo 13% en etapa IV.
En pacientes menores de 3 años con enfermedad localizada la asociación de virilización aislada, presión arterial normal, tumor <200 gramos, totalmente resecado, estadio I y sin ruptura durante la cirugía, tienen una alta probabilidad de supervivencia. Sin cirugía, la sobrevida es sólo de seis a nueve meses.
En nuestra paciente la normalización de los niveles de testosterona y de sus precursores posterior a la cirugía indican control tumoral. Sin embargo, y debido al estadio tumoral con permeación vascular, se requiere de seguimiento con perfil hormonal de precursores adrenales ya que la elevación de las hormonas previamente alteradas serviría como biomarcador y sería altamente sugestiva de recurrencia tumoral.
Hay limitada información del tratamiento con mitotane para disminución de los casos que cursan con hipercortisolismo. También se pueden utilizar drogas que inhiben la síntesis de esteroides como el ketoconazol, metirapona, aminoglutamido o etomidato y suplementos de potasio o espironolactona.
En niños con carcinoma adrenal con enfermedad avanzada se ha administrado cisplatino con doxorrubicina y etopósido de forma paliativa y como quimioterapia previa a recidiva tumoral. La radioterapia no ha demostrado beneficio ni aumento en la supervivencia y, al contrario, puede aumentar el riesgo de segundas neoplasias (sarcoma, etcétera).
Llaman la atención en este caso los antecedentes familiares en la rama paterna de cáncer en segundo y tercer grado, lo que nos obliga a descartar el síndrome de Li Fraumeni que se caracteriza por un patrón de herencia autosómico dominante y la aparición tumoral en edades tempranas de tejidos blandos, sarcomas de hueso, cáncer de mama, carcinoma de corteza adrenal, tumores en el sistema nervioso central y leucemias.
Las mutaciones en la línea germinal del gen supresor tumoral p53 (TP53) son casi siempre el factor predisponente para desarrollar carcinoma suprarrenal. El 70% de los casos de síndrome de Li Fraumeni tiene mutaciones del gen TP53 localizado en el cromosoma 17p13.1 que codifica la proteína p53. Más de 90% de los niños menores de 4 años con carcinoma adrenal presenta una mutación del gen TP53, por lo que el servicio de genética propone realizar el estudio molecular de p53 en esta paciente.

