











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkPRESENTACIÓN
Se presenta el caso de un niño con histiocitosis de células de Langerhans que inicialmente se diagnosticó erróneamente y se le trató quirúrgicamente sin la evaluación integra del paciente.
HISTORIA CLÍNICA
Niño de 7 años, originario de San Luis de la Paz, Gto., con antecedente de otorrea izquierda en una ocasión. Madre de 28 años, escolaridad secundaria, sana; padre de 31 años de edad, escolaridad secundaria, albañil, sano. Tía abuela materna con cáncer de mama, tío con distrofia muscular y tía abuela paterna con cáncer no especificado. Vacunación incompleta.
Inició a los 2 años de edad con alteración de la marcha, pérdida de la fuerza en el pie derecho, caídas al caminar y fue referido a un neurólogo quien solicitó "estudios por alteración neuromuscular". Posteriormente, se agregó dolor intenso al tacto. Acudió a un médico quién diagnosticó osteosarcoma de células gigantes en fémur proximal derecho.
En diciembre de 2009 el paciente sufrió una fractura patológica que se trató en forma conservadora. En 2010 una nueva fractura en el mismo sitio que requirió tratamiento quirúrgico. Hasta antes de su ingreso al Instituto Nacional de Pediatría se realizaron ocho cirugías, la última con colocación de injerto óseo. Una serie ósea realizada fuera del instituto mostró lesiones en sacabocado en radio izquierdo y tibia derecha, además de material de osteosíntesis en fémur proximal derecho.
La exploración física a su ingreso mostró: peso 21 kg (P50-75), talla 98 cm (P10-25), proptosis OD; implantación baja de pabellones auriculares, puente nasal ancho, tabique nasal largo, extremidad inferior derecha dolorosa a la palpación, con cicatriz quirúrgica hipertrófica y salida de material seroso no fétido en región proximal de aproximadamente 5 mm, llenado capilar 2 segundos. Se diagnosticó fístula osteocutánea activa en muslo. Se revisó la serie ósea que mostró lesiones líticas en la tibia derecha, el cúbito izquierdo y la falange proximal del quinto dedo de la mano derecha. Se diagnosticó histiocitosis de células de Langerhans.
Desde tres años antes los padres notaron polidipsia, con ingesta de 5 a 6 litros por día, poliuria, orina clara. Durante su hospitalización se hallaron balances negativos, polidipsia, densidades urinarias con hipostenuria (1002-1004 mOsm/L), hipernatremia (151 mEq/L) y diuresis por encima de 175 mL/m2/h. Se le trató con desmopresina, 44.5 mg, con respuesta de mejoría de la densidad urinaria (1012), disminución de la diuresis (74 mL/m2/h) y de la sed.
La tomografía por emisión de positrones mostró lesiones líticas en región parietal derecha, occipital derecha, cúbito izquierdo, quinto arco costal derecho, pelvis, fémur derecho y tibia derecha. Una tomografía axial computada con destrucción de mastoides, no había lesiones en pulmón. Una resonancia magnética nuclear revelo infiltración a sistema nervioso central, plexos agrandados, silla turca vacía e infundibulitis. Una biopsia de médula ósea fue negativa para neoplasia.
Se le trató con quimioterapia a base de metotrexate, vinblastina y prednisona. Mostró disminución de la proptosis OD y mejoría en el dolor de la pierna derecha. Tuvo crisis convulsivas caracterizadas por tensión mandibular con aumento del tono del hemicuerpo derecho seguido de somnolencia, supraversión de ojo derecho, sin pérdida de conocimiento. A su reingreso tenía disnea intensa. Un análisis mostró Na 159 mEq/L por lo que se le trató con desmopresina y levetiracetam 40 mg/kg/ día previa impregnación. Una tomografía de cráneo mostró una lesión hiperintensa en región occipital derecha sugerente de infiltración. Un electroencefalograma mostró actividad de base lenta con ondas agudas en la región parietooccipital derecha sin evidencia del estatus epiléptico. Posteriormente, el paciente tuvo dificultad respiratoria (polipnea, aleteo nasal). Se hallaron estertores crepitantes de predominio basal derecho. Radiografía de tórax: infiltrados alveolares de predominio derecho, sugestivos de neumonía de probable etiología bacteriana, que se trataron con penicilina G sódica cristalina 300,000 UI/kg/día con evolución favorable.
No hubo respuesta de la enfermedad del sistema nervioso central después de dos cursos de inducción cada uno con metotrexate 500 mg/ m2 por tres dosis, vinblastina, 6 mg/m2 semanal por seis semanas y prednisona 40 mg/m2 por seis semanas. El niño mostró aumento gradual en peso hasta 26.3 kg, con aumento importante de abdomen por el panículo adiposo, hígado 7cmporpercusióntotal,4×4×3cmdebajode borde costal, sin datos de hepatopatía crónica.
La combinación de tomografía por emisión de positrones con tomografía simple mostró actividad metabólica tumoral con lesiones hepáticas en los segmentos II, IV y V, así como hepatopatía difusa en el resto del parénquima de etiología a determinar, que comparada con el estudio previo sugirió infiltración. Por eso se cambió el esquema de quimioterapia por tratamiento con ara C, vincristina y prednisona.
Tuvo hipotiroidismo central probablemente debido a dislipidemia. No fue posible, inicialmente, ofrecer tratamiento pese a LDL muy alta debido a aumento de enzimas hepáticas. Se le trato con acenocumarina, sin datos de sangrado.
La dislipidemia de origen mixto, con predominio de elevación LDL, tiene alto riesgo aterogénico con varios factores, entre los que están el uso de prednisona, la deficiencia de GH, obesidad multifactorial, etc. Al disminuir las enzimas hepáticas se inició medicamento hipolipemiante con simvastatina y ezetimibe para disminuir la producción e incrementar eliminación de colesterol, además de los cambios de estilo de vida. Se continuo con levotiroxina y desmopresina.
Recibió radioterapia 16 Gy en 10 fracciones a cráneo total. Se irradió cráneo con la finalidad de disminuir carga refractaria a tratamientos y debido a respuesta parcial en IRM se reinició radioterapia con 8 Gy en 5 fracciones. En la evaluación mostró mejoría de las lesiones; sólo persistía actividad en el fémur, por lo que inició tratamiento con ácido zolendrónico, 4 mg por dosis por seis cursos, además de mercaptopurina 50 mg/m2 diarios, metrotrexate 20 mg/m2 cada 3 semanas, vinblastina 6 mg/m2 cada tres semanas, con buena respuesta.
COMENTARIO RADIOLÓGICO
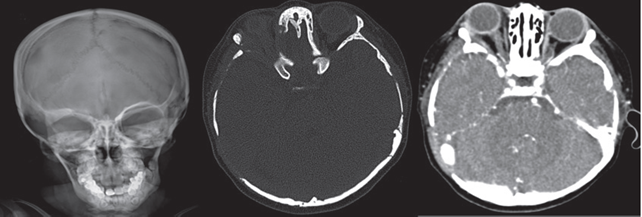
Figura 1 Radiografía simple y tomografía de cráneo con reconstrucciones axiales en ventana ósea; lesiones líticas que involucran el ala menor del esfenoides derecho y los huesos temporal y occipital del lado derecho. La fase contrastada al mismo nivel muestra engrosamiento de tejidos blandos con reforzamiento moderado.

Figura 2 Diáfisis proximal y medial del radio izquierdo con lesión lítica, no expansiva que interrumpe el borde medial de la cortical. Cambios incipientes en el tercio proximal de tibia derecha por áreas de radiolucidez alternantes con radioopacidad.

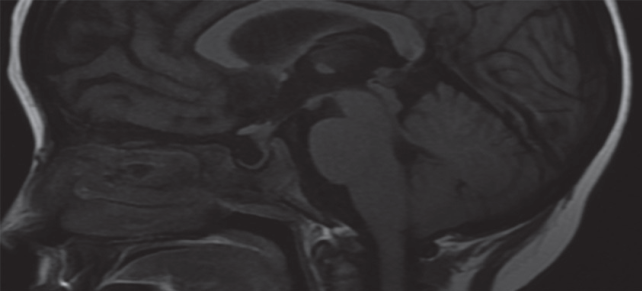
Figura 3 Cortes en secuencia T1 de resonancia magnética con gadolinio que muestran aumento de tamaño de los plexos coroideos, con reforzamiento moderado. En la exploración dirigida a órbitas se logra identificar compromiso del ala menor del esfenoides derecho.
COMENTARIO ONCOLÓGICO
De acuerdo con los órganos afectados y la extensión de la enfermedad se clasifica a la histiocitosis de células de Langerhans en 3 grupos de riesgo:
Enfermedad multisistémica con uno o más órganos de riesgo afectados (hígado, bazo, pulmón y sistema hematopoyético).
De bajo riesgo. Enfermedad multisistémica sin órganos de riesgo afectados.
Enfermedad ósea multifocal y sitios especiales: vertebras con afección de tejido blando intraespinal. Lesiones de sistema nervioso central, órbitas, temporal, esfenoides, cigomático, etmoides, maxilar.
El tratamiento se selecciona de acuerdo con el riesgo. Se han realizado muchos estudios para establecer el mejor tratamiento. En 2013 se publicaron los resultados del estudio HCL III de la Sociedad del Histiocito, se incluyeron más de 400 pacientes para recibir tratamiento a base de vinblastina, prednisona y metotrexate como tratamiento de inducción y un mantenimiento prolongado de 12 meses con los mismos medicamentos además de 6-mercaptopurina. La supervivencia a cinco años fue de 84% con reactivación de la enfermedad hasta en 27% de los casos, mejorando los resultados en comparación con los estudios HCL I y II (62 y 60%, respectivamente).
Nuestro paciente inició su tratamiento con el protocolo HCL III pero no alcanzó una respuesta completa, por ello fue necesario modificar el esquema de quimioterapia y se incluyeron medicamentos como vincristina y ara C, los cuales han dado buenos resultados en pacientes con enfermedad refractaria. Nuestro paciente completó el tratamiento con buena respuesta; no obstante, aun mostraba actividad osteolítica en hueso por lo que se inició tratamiento con bifosfonatos, combinados con vinblastina y prednisona. El empleo de bifosfonatos en pacientes con enfermedad refractaria ha mejorado la supervivencia libre de progresión a tres años de 56.3%, mostrando que es eficaz en el tratamiento de histiocitosis de células de Langerhans con lesiones óseas.
COMENTARIO ANATOMOPATOLÓGICO
En el Departamento de Patología se recibió un bloque de parafina con tejido de inclusión y una laminilla de corte histológico teñida con hematoxilina y eosina enviada del Servicio de Ortopedia con el diagnóstico clínico de probable osteosarcoma. La única información clínica enviada fue que se trataba de una paciente de 7 años con un tumor en fémur distal que fue resecado quirúrgicamente en tres ocasiones.
Ante la ausencia de información sobre la imagen radiológica (si la lesión era única o múltiple y demás características) o si tenía enfermedad sistémica, se analizaron las características histopatológicas de la lesión y posteriormente se hizo la correlación clínico-radiológica.
Histopatología
De manera prominente se observaron en todos los campos numerosas células gigantes multinucleadas xantomatosas entremezcladas con macrófagos espumosos; en menor proporción se encontraron células de Langerhans en forma dispersa y en patrón perivascular, difíciles de reconocer por la evolución crónica de la lesión; sin embargo, éstas estaban acompañadas de ocasionales leucocitos eosinófilos, neutrófilos y linfocitos maduros. Algunas zonas presentaban trayectos de necrosis con cristales de colesterol en un fondo de células gigantes multinucleadas xantomatosas. Otras áreas contenían abundantes neutrófilos y hemosiderófagos, lo que obligaba a descartar una osteomielitis, mientras que en otras se identificaban fenómeno de emperipolesis-like que simulaba enfermedad de Rosai-Dorfman (Figuras 5 y 6).
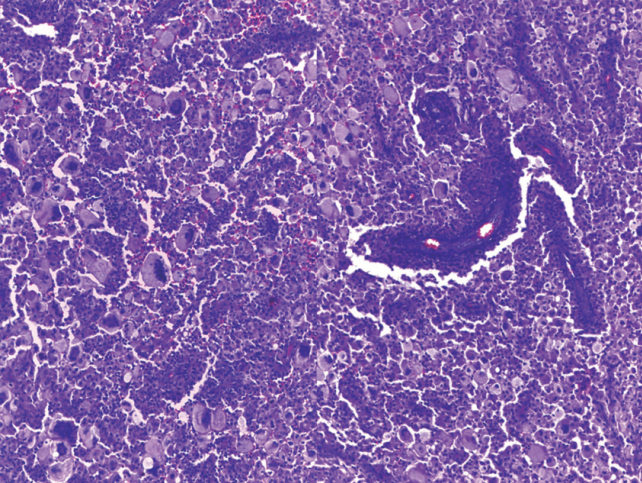
Figura 5 Histiocitosis de células de Langerhans de larga evolución. Varios tipos celulares conforman la lesión: en un fondo de células gigantes xantomatosas y macrófagos espumosos. Se observan células de menor tamaño dispersas y en pequeños cúmulos por toda la lesión y en patrón perivascular.
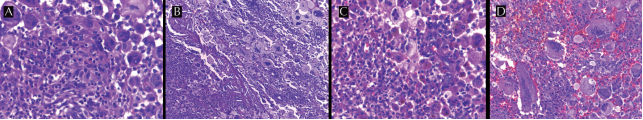
Figura 6 A) Células de Langerhans monoy multinucleadas de citoplasma denso-eosinófilo, núcleos vesiculosos-convolutos y barras nucleares ocasionales; entremezcladas con las células de Langerhans hay linfocitos y leucocitos polimorfonucleares, neutrófilos y eosinófilos B) Trayecto necrótico-eosinófilo que contiene detritus celular y cristales de colesterol. C) Área con predominio de neutrófilos entremezclados con macrófagos y células gigantes multinucleadas; estos últimos exhiben gránulos de hemosiderina en su citoplasma. D) Vista a 40x. Varios tipos celulares íntegros en el citoplasma de las células gigantes multinucleadas, entre ellos linfocitos, polimorfonucleares y macrófagos espumosos.
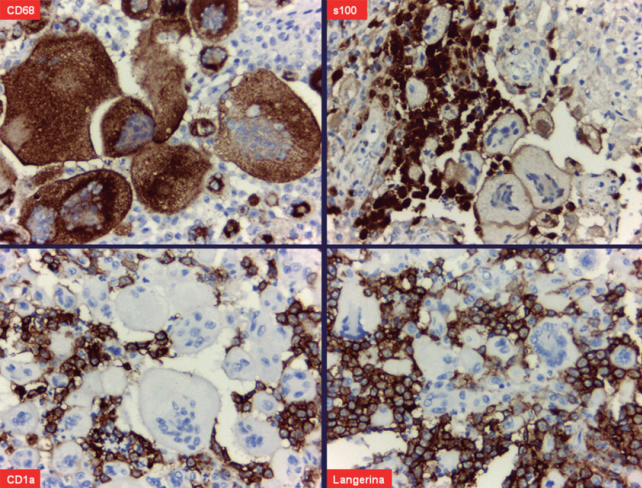
La inmunotinción específica para macrófagos fue intensamente positiva en los macrófagos espumosos y en las células gigantes multinucleadas, con clara negatividad en las células de Langerhans. La proteína s100 (tinción nuclear y citoplásmica) y el CD1a (tinción de membrana citoplásmica) evidenciaron de manera contundente conjuntamente con la langerina, también llamado CD207 (tinción de membrana citoplásmica) la presencia de abundantes células de Langerhans en un fondo xantomatoso (Figura 7).
RESULTADOS
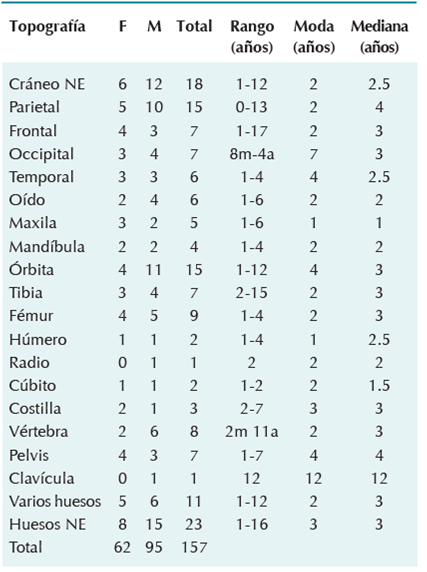
Se encontraron 157 casos de histiocitosis de células de Langerhans ósea en 454 estudios quirúrgicos con diagnóstico histopatológico de histiocitosis de células de Langerhans (35%). De éstos 62 (39%) correspondieron a pacientes femeninos y 95 (61%) al género masculino; con una relación hombre-mujer de 1.5:1 (Cuadros 1 y 2).
Histiocitosis de células monostótica: 146 (93%)
Histiocitosis de células poliostótica: 11 (7%)
Histiocitosis de células unisistema: 147 (94%)
Histiocitosis de células multisistémica: 10 (6%)
Cuadro 1 Histiocitosis de células de Langerhans ósea; archivo del Departamento de Patología (04-08-1971 al 06-032014)

DISCUSIÓN
La histiocitosis de células de una lesión que puede contener abundantes cantidades de células gigantes y éstas pueden ensombrecer a las células de Langerhans. En una histiocitosis de células de Langerhans "madura o antigua" el número de eosinófilos y células de Langerhans pueden estar reducido y sustituido por histiocitos espumosos, que dan a la lesión un aspecto xantomatoso. Además, un fondo de otras células inflamatorias se presenta con frecuencia, incluyendo linfocitos, células plasmáticas, histiocitos y células gigantes multinucleadas, de manera que se puede diagnosticar erróneamente como osteomielitis subaguda o crónica. Sin embargo, en medio de este infiltrado polimorfo se pueden identificar pequeños grupos de células mononucleares de tinción pálida con los característicos núcleos reniformes que muestran ranuras longitudinales. Por otro lado, la mayoría de las lesiones óseas en histiocitosis de células de Langerhans se componen de láminas densas o colecciones discohesivas de células de Langerhans con eosinófilos y células gigantes multinucleadas de tipo osteoclasto interpuestas en el fondo, pero no necesariamente en todos los campos microscópicos de gran aumento.
La necrosis y/o colecciones de eosinófilos e incluso con cristales de Charcot-Leyden pueden dificultar el diagnóstico histopatológico, pero debe sugerir la posibilidad de histiocitosis de células de Langerhans como uno de los intentos para correlacionar las características histológicas y la imagen radiográfica. Las células de Langerhans son confirmadas como tales con la inmunotinción de membrana para CD1a; las células gigantes multinucleadas e histiocitos espumosos no se tiñen con CD1a, pero exhiben inmunotinción citoplásmica para CD68. La inmunotinción de las células de Langerhans para la proteína s100 puede producir una reacción de intensidad variable.
El número y la distribución de las lesiones óseas en histiocitosis de células de Langerhans varían con la edad. En 40% de los niños menores de 5 años de edad, el cráneo, incluyendo el hueso temporal y la mandíbula, están involucrados, mientras que los niños de mayor edad y adolescentes están más propensos a tener una lesión lítica solitaria en un hueso largo (fémur o húmero), cuerpo vertebral, costilla o hueco pélvico. Un cuerpo vertebral puede estar tan amplio o severamente comprimido que se asemeja a una oblea (vértebra plana). Una característica de la histiocitosis de células de Langerhans es que los sitios esqueléticos involucrados, incluyendo las vértebras, se pueden reconstruir. Los niños menores de 2 años de edad son más propensos a tener enfermedad multisistémica con un pronóstico potencialmente grave; por el contrario, los niños mayores pueden tener enfermedad poliostótica y diabetes insípida en 15 a 25% de los casos. El curso clínico general en niños mayores de 5 años de edad es generalmente autolimitado, pero hay excepciones a esto.
CONCLUSIÓN
La presencia de histiocitos xantomatosos puede representar una histiocitosis de células de Langerhans involucionada sin células de Langerhans residuales o representar las características principales de un xantogranuloma juvenil. Una enfermedad de Erdheim-Chester, enfermedad de Rosai-Dorfman u osteomielitis subaguda-crónica. Si ninguno de estos trastornos histiocíticos primarios o un proceso inflamatorio crónico ha sido excluido, la interpretación por defecto es xantoma. Las células xantomatosas también pueden ser prominentes en un fibroma no osificante, displasia fibrosa y en un sarcoma de Ewing-tumor neuroectodérmico primitivo postratado, por lo que este artículo hace un llamado de atención para exigir de forma obligatoria un esfuerzo continuo de todo el equipo médico responsable de los pacientes para establecer correlación clínico-radiológica-histopatológica estrictas de todas las lesiones óseas, para asegurar diagnóstico certero y oportuno antes de aplicar cualquier tipo de tratamiento, cuanto más aquellos que sean altamente invasivos, ya que en muchos hospitales del país los patólogos no cuentan con el apoyo de inmunohistoquímica, la cual en este caso era de vital importancia para el diagnóstico de histiocitosis de células de Langerhans de larga evolución (envejecida).
Además, este caso muestra la necesidad de un tratamiento multidisciplinario adecuado del paciente, por lo que debe ser referido en forma oportuna a instituciones de tercer nivel de atención para su cuidado y seguimiento posterior, lo cual mejorará la posibilidad de supervivencia y disminuirá las secuelas.

