











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkCASO CLÍNICO
Paciente masculino de 5 meses, producto de la segunda gesta de una madre de 33 años, sana, con control prenatal, ingesta de ácido fólico y multivitamínicos. Fue obtenido por cesárea por falta de progresión del trabajo de parto, a término, con peso de 3 800 g, talla 52 cm, Apgar 8-9, lloró y respiró al nacer. Egresó al segundo día como "binomio" sano. Onfalorrexis a los 20 días. Sin antecedentes herodofamiliares de importancia para el padecimiento actual. Alimentado con leche materna exclusivamente hasta el tercer mes; posteriormente recibió alimentación mixta con fórmula de inicio, sin alimentación complementaria. El tamizado metabólico neonatal se reportó como normal, esquema de vacunación completo para la edad.
Padecimiento: inició al nacimiento con distensión abdominal; a los 5 meses la madre notó aumento de la trama vascular abdominal y mayor distensión, apareció ictericia en escleras. Dos semanas después se agregó dificultad respiratoria, acudió al médico que indicó ultrasonograma abdominal que mostró hepatoesplenomegalia y ascitis. Se le refirió al Instituto Nacional de Pediatría.
Exploración física: peso 7 950 g (p 75), talla 62 cm (P 10), perímetro cefálico 39 cm, perímetro abdominal 50 cm, frecuencia cardíaca 148 latidos por minuto, frecuencia respiratoria 40 respiraciones por minuto, tensión arterial 90/50 mmHg, temperatura 37 °C. Indice de Kanawati McClaren con desnutrición moderada, palidez de tegumentos; bien hidratado, normo-céfalo, fontanela anterior normotensa, amplia y posterior cerrada; sin sostén cefálico, ojos simétricos con pupilas isocoricas normorreflécticas, ictericia conjuntival; cuello sin alteraciones; cardiopulmonar sin alteraciones, sólo aumento de la frecuencia respiratoria; abdomen globoso a expensas de líquido de ascitis, hígado de 9 cm, percusión total a 3.5-3.5-3.5 cm debajo del reborde costal derecho, de consistencia aumentada; bazo a 4 cm debajo del reborde costal izquierdo; extremidades hipotróficas, hipotóni-cas y con edema; genitales masculinos Tanner I. Fue tratado con furosemida, espironolactona, restricción hídrica, albúmina, ácido ursodesoxi-cólico y vitaminas liposolubles.
Resultados de laboratorio: hemoglobina 10.3 g/dL, leucocitos 17 700/mm3, neutrófilos 32%, linfocitos 53%, monocitos 10%, eosinó-filos 4%, plaquetas 146 000/mm3, velocidad de sedimentación globular 8 mm/h, tiempo de protrombina (TP) 32" a 22% de actividad, tiempo de tromboplastina parcial (TPT) 34", índice normalizado internacional (INR) 2.72, proteína C reactiva 0.9 mg/dL, albúmina 1.8 g/dL, aspartato amino-transferasa (AST) 496 mg/dL, alanina amino-transferasa (ALT) 203 mg/dL, gam-ma-glutamil transpeptidasa (GGT) 111 mg/dL, fosfatasa alcalina 568 mg/dL, bilirrubina total 11. 2 mg/dL, bilirrubina directa 6.36 mg/dL, bilirrubina indirecta 5.28 mg/dL, creatinina 0.16 mg/dL, Na 129, K 4.3 Cl 105, Ca 8.2, P 2.8, glucosa 90 mg/dL, amonio 53 mMol/L.
Se consideró paciente con hepatopatía crónica descompensada con resultados posteriores de alfa 1 antitripsina, tamizado metabólico ampliado (perfil de aminoácidos, acilcarnitina, ácidos orgánicos, succinilacetona), perfil TORCH, anticuerpos LKM-1, ANA y anti-SMA normales o negativos. Ultrasonido con hepatoesplenome-galia y ascitis, con flujo de la vena porta normal.
Presentó deterioros neurológico y cardiovascular que requirieron intubación y soporte inotrópico con dobutamina y adrenalina; posteriormente paro cardiorrespiratorio en tres ocasiones, revirtiendo a reanimación cardiopulmonar. Inició con hipotermias, por lo cual se agregó tratamiento antimicrobiano. Se reportó crecimiento de bacilo gramnegativo no fermentador en urocultivo por lo que se indicó antibiótico carbapenémico. Ingresó a terapia intensiva con anemia de 6 g/dL, se realizó tomografía axial computada de cráneo que reportó hematoma subdural parietooccipital izquierdo sin efecto de masa. En el abordaje multidisciplinario se reportaron los siguientes hallazgos:
Fondo de ojo: sano ocular sin datos de patología metabólica.
Genética: puntuación 172 índice de sospecha para Niemann Pick tipo C, muestra de mucosa oral negativa.
Neurología: tomografía y resonancia magnética cerebrales que identificaron atrofia córtico-sub-cortical; sin datos compatibles de enfermedad lisosomal.
Electroencefalograma: en vigilia con lentifica-ción generalizada delta. En sueño, actividad mixta delta, theta y beta generalizada de mayor amplitud en regiones parietal y occipital bilateral.
Rehabilitación: deficiencia grave musculoes-quelética.
Evolución: presentó sangrado a través de la cánula orotraqueal, continuó con alteraciones hemodinámicas y tiempos de coagulación prolongados; se agregó neumonía nosocomial, aumento de gasto fecal. Tuvo paro cardiorrespi-ratorio sin responder a la reanimación.
COMENTARIO ANATOMOPATOLÓGICO
Dr. Eduardo López Corella
Al fallecer el niño lo recibimos en el Departamento de Anatomía Patológica con los diagnósticos clínicos finales de neumonía nosocomial, insuficiencia hepática y hepatopatía crónica desconocida.
La autorización en este caso limitaba el estudio post mortem al examen de hígado y a la exploración de la cavidad abdominal. El niño estaba muy ictérico, el abdomen estaba marcadamente distendido, con una red venosa colateral evidente y en la cavidad se encontraron 1 500 mL de líquido de ascitis (Figura 1). Se constató la esplenomegalia y se vio la vesícula un poco chica pero no hipoplásica. El hígado era de aspecto cirrótico, duro y nodular, llamó la atención un prominente nódulo de 2.2 cm de diámetro sobre el lóbulo izquierdo (Figura 2). Se tomó un fragmento grande del hígado que incluía casi todo el lóbulo izquierdo con el nódulo descrito y se tomaron, además, fragmentos pequeños de bazo y de riñón. Los riñones parecían de aspecto normal.

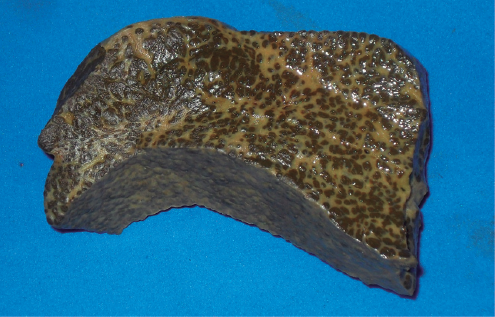
Figura 2 Hígado. En la superficie de corte se aprecia fibrótico y trabeculado con la expansión nodular en parte superior de la pieza. En la parte inferior se aprecia la cápsula con una superficie granular.
El examen del hígado mostró un cuadro mixto de cirrosis obstructiva con colestasis acentuada y una fibrosis de bandas gruesas y limpias, con hiperplasia hamartomatosa de conductos (Figura 3) con un patrón que recordaba a la fibrosis hepática congénita; condición, sin embargo, en la que no suele haber colestasis ni insuficiencia hepática. El fragmento de hígado resecado no incluyó el hilio pero en uno de los cortes se apreció un conducto biliar mayor dilatado y sin epitelio pero con contenido biliar (Figura 4). Este se encontró en un espacio porta mayor y nos sugirió la posibilidad de una enfermedad de Caroli, que sería una malformación de la placa ductal con colestasis.1-3 Como existió evidencia de que no hubo obstrucción biliar extrahepática -la vesícula presente y las heces estuvieron pigmentadas- este caso pareció corresponder a una malformación de la placa ductal con componentes de enfermedad de Caroli y de fibrosis hepática congénita; para esta combinación de dos expresiones de malformación de la placa ductal se ha propuesto el nombre de síndrome de Caroli.4 Los espacios porta periféricos mostraron colestasis acentuada con cambios inflamatorios incipientes en colangíolos (Figura 5) y en el parénquima hepatocelular marcada colestasis y transformación de células gigantes como respuesta a la obstrucción (Figura 6). El nódulo prominente descrito sobre la superficie del hígado en el corte tuvo más o menos el mismo aspecto que el resto y probablemente fue un foco de regeneración nodular. La presencia de regeneración y los datos de obstrucción biliar nos permiten proponer un componente de cirrosis obstructiva superpuesto a la patología de base que serían la fibrosis hepática congénita y los cambios de Caroli.
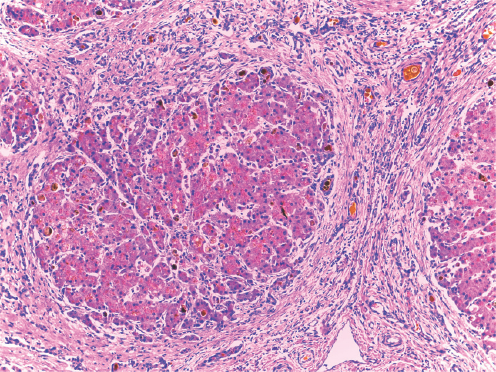
Figura 3 Hígado con bandas fibrosas gruesas y limpias rodeando nódulos de tejido hepátocelular colestáti-co. En el seno de las bandas hay conductos biliares hamartomatosos.
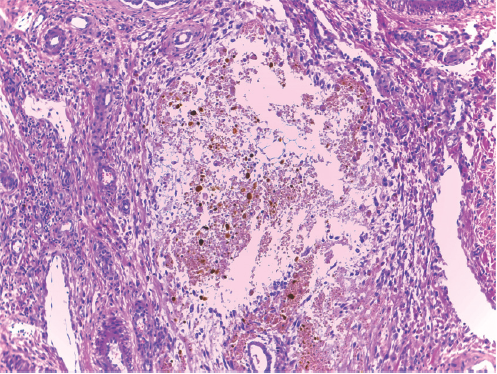
Figura 4 Un conducto biliar mayor dilatado, con retención de contenido biliar, compatible con enfermedad de Caroli.
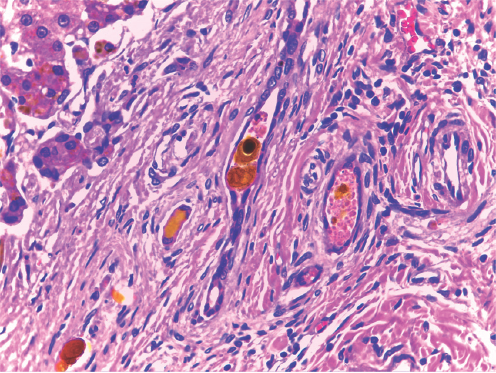
Figura 5 Conductos biliares hamartomatosos con colestasis en el espesor de las bandas fibróticas y con colangitis incipiente.
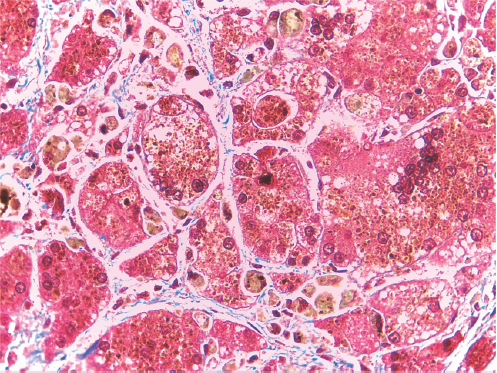
La posibilidad de una patología de la placa ductal obliga a considerar la posibilidad de afección renal que incluye un muy diverso abanico de condiciones que forman parte del espectro de la enfermedad fibroquística hepatorrenal y que pueden asociarse en diferentes combinaciones (Cuadro 1).5 Los riñones no parecían aumentados de tamaño, ni por imagen ni en el examen de la cavidad, pero en el fragmento que tomamos se apreciaron, además de una nefrosis colémica muy acentuada, datos de daño isquémico tubular, túbulos dilatados y glomérulos con dilatación quística del espacio de Bowman (Figura 7); estos cambios, glomeruloquísticos y de ectasia tubular pueden ser indicativos de una enfermedad renal poli-quística de tipo adulto (la enfermedad renal poliquística autosómica dominante) que es parte del espectro reconocido de enfermedades fibroquísticas hepatorrenales que, en cuanto al hígado, parecen ser malformaciones de la placa ductal y tanto en hígado como en riñón muchas de ellas parecen tener como base patogénica un defecto en la función del aparato ciliar y se consideran dentro del capítulo de las ciliopatías.
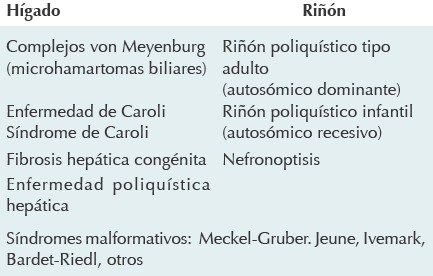
Cuadro 1 Espectro (incompleto) de la enfermedad fibro-quística hepatorrenal. Malformaciones de la placa ductal/ ciliopatías
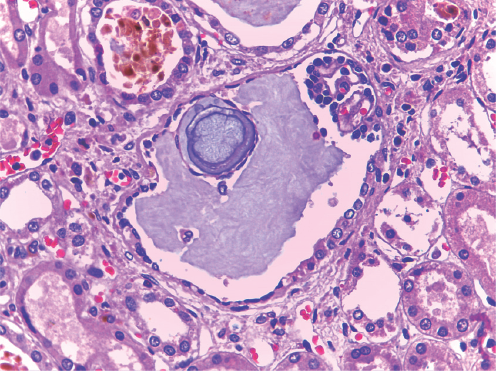
En este paciente se consideró la posibilidad de una enfermedad metabólica. En la autopsia no hubo daño hepatocelular ni esteatosis que apoyaran las posibilidades de galactosemia, tirosinemia ni intolerancia a la fructosa. Tampoco hubo depósitos de material que sugirieran un problema de atesoramiento ni material que oriente hacia deficiencia de alfa-1-antitripsina. Tampoco había acumulación de fierro ni datos que sugieran glucogenosis ni amilopectinosis. La autopsia, si bien parcial, razonablemente descartó una enfermedad metabólica.
Lo que sí parece conveniente en el contexto de consejo genético es el estudio del hermano sano en busca de nefromegalia o alguna alteración sindromática consonante con el espectro de enfermedad fibroquística hepatorrenal que vimos en este paciente.
Una lección que nos deja este caso es que un estudio post mortem, aunque sea parcial, enriquece nuestra visión integral del caso, precisa y expande la información expresada en los diagnósticos clínicos de egreso. Una autopsia completa nos hubiera explicado la patología renal (de la que aquí solo tenemos una pequeña muestra) y nos hubiera revelado la estructura de los elementos de la región del hilio hepático y lo que parece ser una dilatación de los conductos biliares mayores intrahepáticos para así reforzar nuestro diagnóstico de enfermedad de Caroli. Pero aun la autopsia parcial nos aclara el diagnóstico clínico y plantea acciones pendientes de consejo genético.
Diagnósticos anatómicos principales
Enfermedad fibroquística hepatorrenal
Fibrosis hepática con proliferación hamarto-matosa de conductos biliares interlobares y dilatación de conductos intrahepáticos mayores compatible con malformación de la placa duc-tal del tipo síndrome de Caroli (enfermedad de Caroli/fibrosis hepática congénita).
Ectasia tubular renal con cambios glomeruloquísticos
Cirrosis hepática obstructiva con colestasis acentuada y regeneración nodular. (vías biliares extrahepáticas permeables).
Datos anatómicos de hipertensión portal con esplenomegalia congestiva, ascitis y red venosa colateral en abdomen.
Datos de insuficiencia hepática con ictericia y nefrosis colémica.
DISCUSIÓN
Dra. Flora Zárate Mondragón
La enfermedad hepática fibroquística se refiere a un conjunto de enfermedades que afectan al hígado, pueden afectar al riñón y en ocasiones a otros órganos como el páncreas. Se caracteriza por distintos grados de fibrosis hepática y de dilataciones del árbol biliar; éstas son originadas por una malformación de la placa ductal.6-8
La formación de la placa ductal fue descrita por primera vez por Hammar, en 1926, como una parte importante del desarrollo embriológico de las vías biliares. Posteriormente, Jorgensen propuso que la formación de quistes en el árbol biliar era secundaria a una malformación de ésta. Desmet propuso que la persistencia o alteración en el proceso progresivo de remodelación de la placa ductal origina un espectro muy grande de diferentes alteraciones en hígado, vías biliares y riñón.5
La malformación de la placa ductal puede estar mediada por alteraciones en los genes que codifican las proteínas que regulan el desarrollo y remodelación de ésta; así como en la señalización de los cilios involucrados en la misma.5,9-11
Las enfermedades que se han asociado con esta malformación son múltiples y el cuadro clínico que presentan es muy variado: asintomáticas, con daño hepático o renal, algunas con insuficiencia renal temprana o tardía, otras con hipertensión portal, colestasis, colangitis, etcétera. Las más importantes son: los complejos de Meyenburg, la fibrosis hepática congénita, la enfermedad poliquística renal autosómica recesiva, la enfermedad poliquística renal auto-sómica dominante, la enfermedad y el síndrome de Caroli, entre otras.6,7,11
La placa ductal es una estructura cilíndrica de doble capa que inicia su formación entre las sexta y séptima semanas de la gestación, es la precursora de los conductos biliares intrahepáticos y de las glándulas peribiliares intrahepáticas. La placa presenta un proceso de remodelación que empieza en la onceava semana (apoptosis y proliferación celular); en dicho proceso intervienen los cilios y su señalización por numerosas moléculas como proteinasas e inhibidores tisula-res de la matriz, metaloproteinasas, tripsinógeno, catepsina, moléculas relacionadas con la apoptosis celular (Bcl-2, Levis Y y C-myc), TGF-α y su receptor, MET, C-erbB2, E-caderina, catetinas (α, β y γ), alfa amilasa pancreática, PKR, apomucina-sas y el plexo capilar peribiliar. Estas alteraciones originan un espectro muy grande de diferentes enfermedades hepáticas y/o renales.5,6,9 Desmet describió que dependiendo del tamaño afectado de los conductos biliares y si existe ectasia o bien involución, será la expresión clínica; de tal forma que si se afectan los conductos mayores se presentará el síndrome de Caroli y la enfermedad de Caroli, de mediano calibre enfermedad poliquística renal autosómica recesiva y fibrosis hepática congénita, de pequeño calibre enfermedad poliquística renal autosómica dominante y complejos de Meyenburg.12,13
Los complejos de Meyenburg son lesiones hamartomatosas biliares que habitualmente se encuentran incidentalmente en un hígado sano. Generalmente son asintomáticas.
La fibrosis hepática congénita puede encontrarse como afección única o asociarse, en la mayoría de los casos, con la enfermedad poliquística renal autosómica recesiva y en ocasiones con nefroptisis; su prevalencia no se conoce. Se han descrito cuatro formas de presentación: hipertensión portal, colangítica con colangitis de repetición, mixta y latente. El cuadro clínico puede presentarse en niños escolares y adolescentes como hipertensión portal sin o con poco daño renal; en estos pacientes puede haber hepatomegalia a expensas del lóbulo izquierdo; el derecho no se logra palpar. En los lactantes habitualmente existe daño renal (enfermedad poliquística renal autosómica recesiva) y posteriormente se asocia hepatomegalia y datos de hipertensión portal. Las pruebas de función hepática en la fibrosis hepática congénita no complicada son típicamente normales o con discreta elevación de las pruebas de inflamación; sin embargo, es común encontrar hiperesplenis-mo que en muchas ocasiones es el motivo de consulta. El ultrasonido hepático mostrará un patrón en parches con ecogenicidad aumentada y esplenomegalia; el Doppler datos de hipertensión portal con aumento de la velocidad de flujo de la vena porta y circulación colateral. La biopsia hepática ofrecerá el diagnóstico como en el caso de este paciente.5,8,11,14
En las ciliopatías el órgano más comúnmente afectado es el riñón. De éstas las más comunes son las enfermedades poliquística renal autosómica recesiva y dominante; el espectro de estas alteraciones incluye también a la nefroptisis, la displasia quística renal, el riñón con médula esponjosa, entre otras.5
La enfermedad poliquística renal autosómica recesiva, previamente conocida como enfermedad poliquística infantil, se estima que ocurre en 1 de cada 6 000-40 000 nacidos vivos. El gen involucrado en esta afección es el PKHD1. La enfermedad poliquística renal autosómica recesiva habitualmente se manifiesta con insuficiencia renal progresiva con múltiples quistes fusiformes no comunicantes y no altera la forma del riñón; sin embargo, sí aumenta su tamaño; esta lesión puede ser aislada, asociarse con la fibrosis hepática congénita o bien con dilatación de la vía biliar y fibrosis hepática congénita (síndrome de Caroli). La enfermedad poliquística renal autosómica recesiva puede ser de aparición neonatal, en el lactante, en niños y en adolescentes; sin embargo, esto no correlaciona con la gravedad. La mayoría de los pacientes que se presentan con falla renal en etapas tempranas de la vida requerirá trasplante renal en la etapa adulta. En 80% de los casos los niños pueden presentar hipertensión arterial de difícil control que requiere tratamiento con varios antihipertensivos; cuando aparece en niños mayores la hipertensión suele ser leve y de fácil control. Eventualmente, cuando se asocia con fibrosis hepática congénita aparecen datos de hipertensión portal, cuando se asocia con el síndrome de Caroli puede haber datos de colangitis con progresión del daño hepático.6,7,11,14
La enfermedad poliquística renal autosómica dominante es una ciliopatía sistémica en la cual el riñón y el hígado están primariamente afectados, pero también puede haber quistes en páncreas, aracnoides y vesículas seminales. Se puede asociar con aneurismas intracraneales, dilatación del arco aórtico, disección de la aorta torácica y prolapso de la válvula mitral. La prevalencia reportada es de 1 en 500-1 000 nacidos vivos. Los genes involucrados son el PKD1 y PKD2. Esta variedad se presenta como insuficiencia renal en la adultez con la mutación de la PKD1; la insuficiencia renal ocurre a la edad de 20 años y con la de la PKD2 en la década de los 50. La mayoría de los pacientes nace con riñones normales, los quistes se presentan y crecen a lo largo de la vida: 58% entre los 15 y 24 años de edad y 94% entre los 35 y 46 años. La función glomerular empieza a declinar y se desarrolla hipertensión arterial conforme crecen los quistes. En niños puede encontrarse un quiste unilateral pero debe darse seguimiento por la probable progresión, sobre todo en el caso de que existan familiares afectados.6,7,13
La enfermedad de Caroli y el síndrome de Caroli deben su nombre a que Caroli describió dos formas de dilataciones biliares intrahepáticas congénitas. El síndrome de Caroli es el más frecuente; se caracteriza por la presencia de fibrosis hepática congénita y dilatación del árbol biliar. La enfermedad de Caroli sólo se caracteriza por la presencia de ectasia de los conductillos biliares. Estas anormalidades son más comunes en adultos; sin embargo, se pueden presentar en niños; son trasmitidas por herencia autosómica recesiva y frecuentemente se asocian con enfermedad poliquística renal autosómica recesiva y rara vez con la forma dominante. Se ha postulado que la enfermedad de Caroli es secundaria a una alteración de los conductos biliares grandes y por eso se le asocia con el quiste de colédoco e inclusive se le incluye en su clasificación, punto actualmente controvertido. El síndrome de Caroli se asocia con una alteración de la formación de todo el árbol biliar, incluyendo los conductos biliares interlobulares. Como demostró el estudio histopatológico de nuestro paciente, él correspondió a esta variedad de alteración de la placa ductal. En estas afecciones la malformación de los conductos biliares predispone a la estasis biliar, lo que puede ocasionar cuadros de colan-gitis con daño hepático progresivo (cirrosis de tipo biliar) como en el caso de nuestro paciente. La estasis puede producir colelitiasis, colangitis, abscesos biliares, septicemia y, en la edad adulta, se asocia con colangiocarcinoma.6,15
Habitualmente el ultrasonido hepático muestra dilatación de la vía biliar y el patrón ecogénico alterado por la fibrosis hepática congénita.11 En este paciente llama la atención que el ultrasonido hepático no reveló dilatación de los conductos biliares intrahepáticos; tampoco en el Doppler se demostró hipertensión portal.
Este caso se presentó como una hepatopatía crónica con insuficiencia hepática, sin dilatación o quistes en la vía biliar o renal, por lo que fue necesario abordarlo como tal y por ello se descartaron enfermedades infecciosas, metabólicas y autoinmunitarias. Sin embargo, por las condiciones del paciente no se logró realizar la biopsia hepática que, como hemos visto, es la que nos ofrecería el diagnóstico.

