











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkRESUMEN CLÍNICO
Niño de 4 años de edad con diagnóstico de deficiencia de adhesión leucocitaria tipo 1, postrasplantado de células progenitoras hematopoyéticas en tres ocasiones, las dos primeras con falla primaria de injerto. Producto de segunda gesta con adecuado control prenatal. Obtenido por cesárea porque la mamá tuvo placenta acreta y preeclampsia. Al nacer tuvo un peso de 3 800 g, Apgar 8-8, egresando en binomio conjunto. Esquema de vacunación de acuerdo con el implementado en Estados Unidos por lo que no recibió inmunización para tuberculosis. Antecedentes heredofamiliares de importancia: hermana finada a los seis meses de edad por absceso umbilical, síndrome disentérico y abscesos perianales. Además, medio hermano con síndrome Prader Willi.
A los 11 días de vida inició su padecimiento con onfalitis que fue tratada con penicilina, ceftriaxona y ceftibuteno; remisión parcial. Al mes de vida ingresó por primera vez a nuestra institución por otorrea bilateral y ataque al estado general. Se encontraba en tratamiento antimicrobiano con ceftriaxona y, días antes, había presentado onfalorrexis. Dados los antecedentes se decidió hospitalización e inicio de abordaje diagnóstico por sospecha de ¡nmunodeficiencia primaria. Se realizó bio-metría hemática: Hb 8 g/dL, Hct 23, leucocitos 22 100 cel/mL, neutrófilos 12 155 cel/mL, linfocitos 6 630 cel/mL, monocitos 1 768 cel/mL, plaquetas 606 000, bandas 4%. Se reportaron: inmunoglobulina A 254 mg/dL; inmunoglobulina G 552 mg/dL; inmunoglobulina M 87 mg/dL; fracción C3 del complemento 149 mg/dL; fracción C4 del complemento 13.5 mg/dL; subpoblaciones de linfocitos con linfocitos T (CD3+) 13 840 cel/ mm3 (85%); linfocitos T cooperadores (CD4+) 10 986 cel/mm3 (85%); linfocitos T citotóxicos (CD8+) 2 089 cel/mm3 (15.54%), (CD16+56+) 1 373 cel/mm3 (8.7%). Se realizó citometría de flujo con expresión de CD11b en linfocitos 0.28%, en granulocitos 0.03%, en monocitos 0.58%, CD18 en linfocitos 0.32%, en granulocitos 0.04%, en monocitos 0.15%. Por los antecedentes de la hermana fallecida referidos más arriba y los antecedentes de onfalorrexis tardía, así como disminución de CD11 y CD18, se sospechó deficiencia de adhesión leucocitaria tipo 1.
Durante sus primeros dos años de vida se hospitalizó en seis ocasiones por causas infecciosas en las que predominaron cuadros gastroenteritis, así como balanopostitis, celulitis perianal y en el tercer dedo de la mano izquierda; recibió tratamiento con antibiótico. Durante los procesos infecciosos llegó a cursar con leucocitosis entre 85 000 a 20 000 cel/mL y neutrofilia.
A los dos años de edad se le realizó el primer trasplante de células progenitoras hematopoyéticas con fuente de células madre (CD34+) derivadas de cordón umbilical y dosis de 4.4 x 105/kg células. Se utilizó régimen de acondicionamiento con busulfano, ciclofosfamida y globulina antitimocito (Cuadro 1). Tuvo falla primaria de injerto complicada con sepsis por Pseudomonas aeruginosa relacionada con el catéter venoso central; fue tratada con ceftazidima y amikacina.
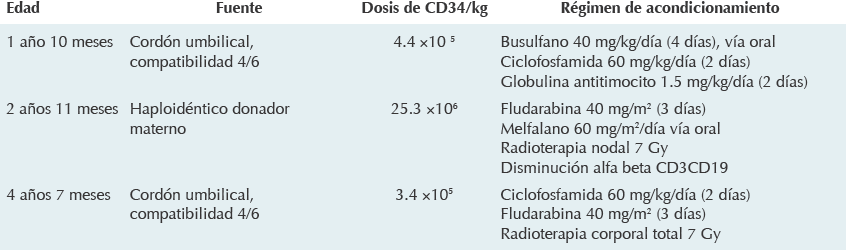
Después de la falla en el primer trasplante continuó con cuadros infecciosos, predominando el de gastroenteritis, así como infecciones de los tejidos blandos y requiriendo hospitalización en múltiples ocasiones. Se decidió realizar un segundo trasplante de células progenitoras hematopoyéticas que fue haploidéntico donador materno con dosis celular CD34+ de 25.3 x 106/kg con régimen de acondicionamiento con base en fludarabina, carboplatino y radioterapia nodal 7 Gy; nuevamente hubo falla primaria de injerto.
Después del segundo trasplante el paciente cursó con anemia hemolítica, púrpura trombocitopéni-ca autoinmunitaria y se detectaron anticuerpos anti-HLA clases I y II, por ello se le administraron cuatro dosis de rituximab logrando negativizar los anticuerpos anti-HLA (Cuadro 1).
El paciente continuó con cuadros de gastroenteritis infecciosa, abscesos perianales recurrentes y un cuadro de dolor abdominal que evolucionó a perforación intestinal; requirió laparotomía exploradora con hemicolectomía derecha, se complicó con infección de la herida quirúrgica e infección asociada al catéter. Después tuvo plastrón abdominal con formación de absceso y choque séptico, requirió una segunda intervención quirúrgica con resección de íleon de 15 cm y ameritó estancia en terapia intensiva con manejo ventilatorio y tratamiento aminérgico. Mejoró pero continuó hospitalizado durante un año por cuadros infecciosos recurrentes que requerían tratamiento antibiótico intravenoso de amplio espectro.
Ante la persistencia de las infecciones que ponían en peligro su vida y la existencia de una fuente de células progenitoras hematopoyéticas compatibles se decidió realizar un tercer trasplante a la edad de 4 años 7 meses (Cuadro 1). A las 24 horas de infusión de las células el paciente cursó con fiebre persistente y sepsis grave; se aisló en hemocultivo Pseudomonas aeruginosa y cocos grampositivos. Además se encontró celulitis en el dorso de la pierna, de rápida progresión, que fue tratada con linezo-lida (10 mg/kg/d), meropenem (20 mg/kg/do), caspofungina (50 mg/m2) y teicoplanina (6 mg/ kg/d). El cuadro evolucionó a choque séptico en 48 horas. Hubo paro cardiorrespiratorio que no se revirtió con las maniobras avanzadas de reanimación.
HALLAZGOS ANATOMOPATOLÓGICOS
El tejido obtenido de la laparotomía exploradora por perforación intestinal correspondió a 16 cm de intestino conformados por: íleon terminal, válvula ileocecal y colon ascendente. La superfìcie externa presentó aspecto violáceo con un plastrón anfractuoso y rojizo en la región retrocecal; en los cortes histológicos este plastrón correspondía a una perforación extensa de la pared apendicular con exudado fibrinoso, carecía de neutrófilos y sólo contenía algunos macrófagos. En la pared intestinal conservada del íleon terminal y colon ascendente no hubo tejido linfoide (Figuras 1 y 2). Posteriormente se recibieron, en el Departamento de Patología, un fragmento de 20 cm de colon transverso con una perforación de 3 cm y otro fragmento de 5 cm de íleon con los mismos hallazgos histopatológicos.
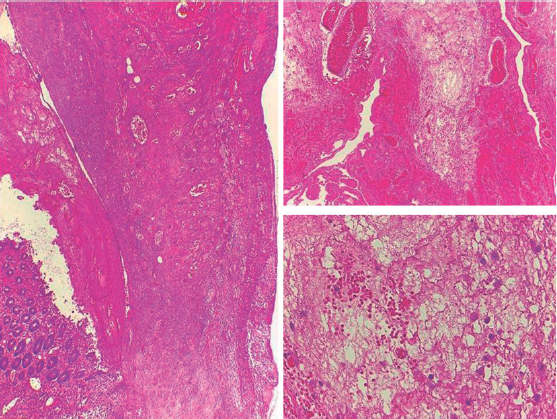
Figura 1 Perforación de la pared apendicular. A) Escaso epitelio residual que se continúa con destrucción extensa de la pared asociado con hemorragia reciente y exudado fibrinoso. B) Aumento a mediano poder para evidenciar zonas de hemorragia reciente que alternan con exudado fibrinoso. C) Acercamiento del exudado fibrinoso: ausencia de leucocitos polimorfo-nucleares, solo se observan macrófagos ocasionales.
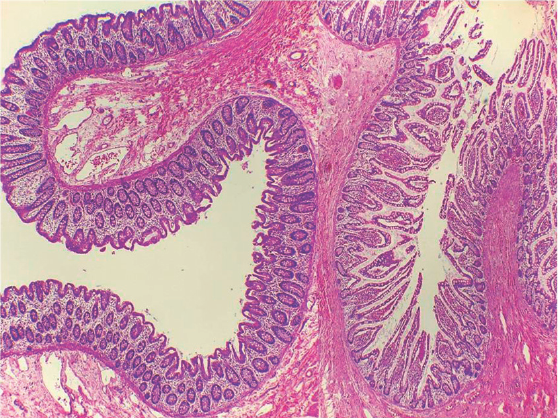
Figura 2 Pared íntegra de íleon terminal, válvula ileocecal y colon ascendente sin evidencia de tejido linfoide.
Quince meses después de la última resección se realizó autopsia completa a las 7 horas del fallecimiento. Los hallazgos anatomopatológicos consistían en hipotrofia e hipoplasia extrema del timo, hipoplasia linfoide grave generalizada, hepatoesplenomegalia y nefromegalia congestivas (Figura 3). La condición que causó la muerte fue un choque séptico por Pseudomonas aeruginosa corroborado histológicamente y por bacteriología post mortem (Figura 4).
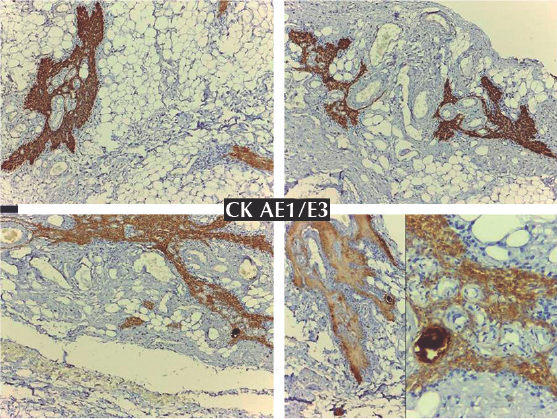
Figura 3 Lóbulos tímicos residuales inmunoteñidos con CKAE1/AE3, se demuestran células epiteliales corticales subcapsulares e internas, así como células epiteliales medulares y células epiteliales de corpúsculos de Hasssall; nótese la despoblación global de tejido linfoide y el aumento consecuente de el tejido fibroadiposo.

DISCUSIÓN
En el abordaje de una inmunodeficiencia primaria onfalorrexis tardía, onfalitis, infecciones graves y leucocitosis hacen sospechar, en general, de un defecto de la fagocitosis, y en particular en deficiencia de adhesión de leucocitos tipo 1.1 Esta inmunodeficiencia primaria se puede clasificar en tres grupos. En la tipo 1 existe defecto en la adhesión de los leucocitos al endotelio debido a mutaciones en el gen (ITGB2) que codifica para la subunidad beta 2 integrina (CD18), esencial para las conexiones con las células tisulares o las proteínas de la matriz extracelular para lograr la movilización de los leucocitos.2,3 Cada beta 2 integrina es un heterodímero compuesto por una cadena alfa (CD11a, CD11b, CD11c o CD11d) unida, no covalente, a la cadena beta 2 (CD18), la asociación de ambas subunidades es necesaria para una expresión y función normales.4 En nuestro paciente ésta fue la inmunodeficiencia diagnosticada y los hallazgos característicos de laboratorio fueron leucocitosis a expensas de neutrófilos además de expresiones disminuidas, por citometría de flujo, de CD18 y CD11b (< 2%).3
Una de las manifestaciones clínicas más representativas de esta entidad son las infecciones en los tejidos blandos (que pueden ser indoloras, necróticas y con riesgo de recurrencia) asociadas con bacteriemias secundarias; datos que presentó nuestro paciente. Debido al defecto para la adhesión de neutrófilos en los sitios de lesión, por lo general, no se documenta pus. Es importante mencionar que estos hallazgos se pudieron corroborar en las biopsias de tejido apendicular (Figura 1) donde no se observaron polimorfonucleares, células típicas en una infección, por lo que estos pacientes pueden evolucionar lentamente (de manera insidiosa) y ocasionar perforación intestinal sin tener todos los datos clínicos de ésta; ante cualquier indicio de manifestaciones se debe iniciar la cobertura antibiótica o el tratamiento quirúrgico.3
Se ha correlacionado una menor expresión de CD18 (< 2%) con un fenotipo más grave y esto es indicativo de trasplante de células progenitoras hematopoyéticas. Los pacientes con deficiencia de adhesión de leucocitos tipo 1 grave tienen un pronóstico sombrío y fallecen antes de los dos años por infección;5 el trasplante es el único tratamiento curativo. Se ha reportado una tasa de supervivencia de hasta 75% con donadores 100% compatibles.2,6
Antes del trasplante de células progenitoras hematopoyéticas los tratamientos descritos son: utilización de antibióticos de amplio espectro en caso de infección; administración de antibióticos profilácticos: trimetoprim con sulfametoxazol e itraconazol, así como tener un buen estado nutricional. Se ha reportado también el uso de transfusiones de granulocitos en caso de infecciones graves como el ectima gangrenoso.7
En el tratamiento de este paciente también se administró inmunoglobulina intravenosa con el fin de mejorar la opsonización de los microorganismos y para prevenir la diseminación del proceso infeccioso; así mismo, se administró gammaglobulina subcutánea local y factor estimulante de colonias de granulocitos con el objetivo de un efecto inmunomodulador local.
Dado el fenotipo tan grave del paciente se decidió realizar trasplante de células progenitoras hematopoyéticas. La mayoría los trasplantes realizados en el mundo son de médula ósea y con disminución de linfocitos T en caso de an-tígeno de histocompatibilidad no idéntico. Entre las principales complicaciones reportadas están las infecciones graves que deben ser tratadas de manera agresiva para obtener el mejor control de cuadros infecciosos residuales previos al procedimiento. En nuestro paciente este fue uno de los factores de difícil control, ya que persistía con cuadros infecciosos durante los trasplantes, lo que pudo ser un factor determinante para la falla del injerto.8
La falla del injerto es un factor importante en la morbilidad y en la mortalidad posteriores al trasplante de células progenitoras hematopoyé-ticas. Se clasifican en primarias y secundarias: las primarias se caracterizan por persistencia de neutropenia (cuenta de neutrófilos totales < 0.5 x 109/L) que provoca pancitopenia y necesidad urgente de retrasplante; las secundarias ocurren por la pérdida de células del donador después de un injerto inicial.9
Una teoría a la que también se puede atribuir la falla de injerto en la deficiencia de adhesión de leucocitos tipo 1 es que en estos pacientes se ha demostrado que las células del linaje mieloide macrofágico están sobreactivadas, por lo que requieren esquemas de acondicionamiento mie-loablativo agresivo para poder superar el riesgo de rechazo, así como para facilitar su injerto. Dado que en los pacientes pediátricos existe disminución de la biodisponibilidad del busul-fano, que es requerido en estos esquemas, no se puede garantizar totalmente su efecto.8 La administración de esquemas mieloablativos puede tener como efecto secundario mayor toxicidad e incremento del riesgo de infecciones. Además, se ha reportado un número basal incrementado de células progenitoras, en la médula ósea de los pacientes con deficiencia de adhesión de leucocitos, ocasionado por una estimulación crónica relacionada con infecciones bacterianas persistentes.10,11 Todos estos factores pudieron condicionara la falla de injerto en el trasplante de nuestro paciente.
Se han descrito también esquemas de acondicionamiento de intensidad reducida que pueden ser suficientes para el establecimiento de una adecuada reconstitución inmunológica con menos toxicidad.12
Los esquemas que se utilizaron en nuestro paciente fueron: en el primer trasplante de células progenitoras hematopoyéticas un esquema mieloablativo con el fin de garantizar un injerto adecuado. Un punto importante a considerar es que dada la disponibilidad de fármacos se utilizó busulfano vía oral; sin embargo, existen reportes de mayor biodisponibilidad de manera intravenosa que puede contribuir a disminuir el porcentaje de éxito. El segundo esquema fue de intensidad reducida y con disminución de linfocitos T para menor toxicidad y mejor posibilidad de injerto. Finalmente, en el tercer trasplante se utilizó nuevamente como fuente el cordón umbilical con esquema de intensidad reducida.
Un aspecto importante a considerar en el trasplante de células progenitoras hematopoyéticas de esta inmunodeficiencia primaria es que se han descrito buenos resultados con quimeris-mos mixtos y que la corrección clínica de la enfermedad se ha reportado entre 7 y 30% de las células del donador; esto también se puede vigilar mediante la medición de la expresión de CD18/CD11 por citometría de flujo después del trasplante. En nuestro paciente no se tuvieron nuevas determinaciones que demostraran ese grado de injerto.11
Otros hallazgos de importancia en este paciente fueron la hipoplasia o ausencia de tejido linfoide, la hipotrofia e hipoplasia extrema del timo (Figuras 2 y 3). Estos datos son relevantes ya que en este paciente no sólo evidenciamos la deficiencia de neutrófilos sino también el importante compromiso de la inmunidad celular por linfocitos T y B comportándose, de manera indirecta, como inmunodeficiencia combinada grave; estos pudieron ser secundarios a los esquemas de acondicionamiento recibidos, contribuyendo a la persistencia de infecciones y al desenlace fatal.
En un futuro no solamente el trasplante de células progenitoras hematopoyéticas será la opción curativa de esta inmunodeficiencia primaria. Se encuentran en protocolo ensayos para establecer terapia génica dirigida, actualmente con buenos resultados en modelos caninos con corrección y mejoría de la enfermedad sin efectos adversos. Esperaremos los ensayos clínicos en humanos.13
CONCLUSIONES
Debe sospecharse déficit de adhesión leucocitaria ante cuadros de onfalitis, onfalorrexis tardía e infecciones de tejidos blandos, combinadas con leucocitosis y disminución de la expresión de CD18/CD11. Es importante enfatizar que estos pacientes cursan con falla en la adhesión de los neutrófilos a los sitios de infección, por lo que los cuadros pueden ser subclínicos. Se debe tener alta sospecha para tratarlos oportunamente. Las mejores condiciones para el trasplante de células progenitoras hematopoyéticas son: un donador con antígeno de histocompatibilidad idéntico, con remisión o control (al máximo) de los cuadros infecciosos, el mejor estado nutricional y, dependiendo de cada caso, se podrán adecuar los esquemas de acondicionamiento en virtud de garantizar la mayor supervivencia. El diagnóstico oportuno de ésta y de todas las inmunodeficiencias primarias es de vital importancia para prevenir cuadros infecciosos que incrementan el riesgo de falla de injerto en el trasplante de células progenitoras hematopoyéticas y, como consecuencia, un desenlace fatal.

