











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkPRESENTACIÓN DEL CASO
Mujer adolescente, producto de la primera gestación de una madre de 18 años de edad, misma que recibió atención prenatal adecuada y cursó con evolución normal. Se obtuvo a término (a las 38 semanas de gestación) con peso al nacer de 3 400 g. Al interrogatorio se recordó una somatometría neonatal como único dato. Su padecimiento inició al mes de vida con vómitos que generaron deshidratación y hospitalización en unidad de segundo nivel; ésta se prolongó por 5 semanas debido a problemas renales, pulmonares y persistencia de alteraciones metabólicas que finalmente condujeron al diagnóstico de error innato del metabolismo: acidemia metilmalónica.
Acudió inicialmente al Instituto Nacional de Pediatría, a los 4 meses de edad, por un cuadro de tos y rinorrea hialina que no mejoró con tratamiento sintomático; al octavo día se agregó vómito de contenido gástrico e hiporexia. A su ingresó se encontró somnolienta y se documentó acidosis metabólica no compensada, con hiato aniónico incrementado y con hiperamonemia de 253 mg/dL, por lo que se inició tratamiento con medidas antiamonio (rehidratación y bicarbonato).
La evaluación del Departamento de Genética de la Nutrición corroboró el diagnóstico de acidemia metilmalónica y se inició tratamiento con fórmula de aminoácidos libre de metionina, valina y baja en isoleucina y treonina (Propimex 1®), miel de maíz, medidas antiamonio (benzonato de sodio, neomicina), producto 800056, piridoxina y cobalamina. Egresó 10 días después en condiciones estables.
En el seguimiento cursó con múltiples hospitalizaciones por cuadros de descompensación metabólica, en algunas ocasiones con inestabilidad grave, requirió de cuidados intensivos y de ventilación mecánica. En evaluaciones posteriores se agregaron los diagnósticos de retraso grave del neurodesarrollo y acidosis tubular renal, por lo que se inició con rehabilitación neurológica y bicarbonato. También se realizó gastrostomía y fundoplicatura tipo Nissen para facilitar su manejo ambulatorio.
A los 14 años presentó petequias y equimosis en las extremidades superiores que se extendieron a la cara, tórax y pies. Acudió a urgencias de nuestro Instituto por dolor abdominal intenso. A su ingreso, y después de una evaluación inicial, se integró el diagnóstico de abdomen agudo; en la evaluación paraclínica se documentó descompensación metabólica grave (Cuadro 1). Se iniciaron medidas de corrección de amonio e hiperglicemia; sin embargo, la paciente evolucionó mal, tuvo fiebre, taquicardia, llenado capilar lento y datos de insuficiencia respiratoria. Se proporcionó apoyo mecánico a la ventilación, cargas de cristaloide, coloide y apoyo aminérgico sin respuesta. Culminó con paro cardiorrespiratorio y muerte.

COMENTARIO CLÍNICO
La principal afección con la que esta paciente cursó fue una acidemia metilmalónica que, a pesar de una respuesta parcial al tratamiento proporcionado, le generó diversas hospitalizaciones, complicaciones, secuelas y finalmente la muerte. El reconocimiento temprano de un paciente con un error innato del metabolismo es indispensable para ofrecer un tratamiento oportuno pues puede disminuir potencialmente el impacto de la enfermedad sobre la calidad y el pronóstico de vida. Como pediatras debemos conocer los aspectos fisiopatológicos y clínicos que explican el desarrollo de la acidosis metabólica en un paciente para poder identificar su causa y ofrecer una solución.
La acidosis metabólica corresponde a situaciones en las que existe aumento en la producción de hidrogeniones (acidemias orgánicas, ce-toacidosis diabética), administración de ácidos (intoxicación por salicilatos), pérdidas de bicarbonato (diarrea aguda) o incapacidad del riñón para excretar hidrogeniones (acidosis tubular renal).1,2 La acidosis metabólica cursa con un descenso del bicarbonato y del pH, así como aumento de la presión parcial del dióxido de carbono (PCO2), lo que estimula al centro respiratorio y da lugar a un incremento en la frecuencia respiratoria y del volumen corriente compensando con alcalosis respiratoria (i.e. eliminando CO2 con cada espiración logrando disminuir la PCO2). Tisularmente se lleva a cabo un intercambio de potasio intracelular por hidrogeniones séricos, provocando hiperpotasemia. Por otra parte, la acidosis aumenta la producción de amoniaco y la excreción de hidrogeniones (H+) por el riñón.1,2 Clínicamente se acompaña de respiración de Kussmaul (hiperventilación compensatoria), náuseas y vómitos, depresión del sistema nervioso central con somnolencia, crisis convulsivas e incluso coma.1,2 En los cuadros leves el aparato cardiocirculatorio responde con taquicardia y aumento del gasto cardíaco. En las formas graves se produce descenso de la contractilidad miocárdica, con abatimiento de las resistencias periféricas, dando lugar a hipotensión, hipoxia tisular, cianosis y finalmente choque.1,2
En su primer evento la paciente cursó con datos similares a los descritos para la acidosis metabólica: náuseas, vómitos, somnolencia y gasometrías que corroboraron la afección. En el abordaje de un paciente de estas características es importante descartar las causas más frecuentes y de mayor gravedad. En términos generales el interrogatorio inicial permite reconocer si el paciente tiene historia de pérdidas gastrointestinales por vómito o diarrea. Si tiene riesgo de intoxicación por ácidos exógenos o medicamentos que favorezcan acidosis metabólica como ácido acetilsalicílico, etilenglicol, metanol, metformina o linezolida, entre otros; si existe historia sugestiva de diabetes mellitus y riesgo de cetoacidosis o si hubo algún factor desencadenante claramente identificado.
El segundo aspecto importante es la clasificación de la acidosis metabólica de acuerdo con el hiato aniónico documentado en una gasometría arterial, ya sea éste normal o aumentado. Esto permite identificar si se trata de pérdida de bicarbonato o incremento en la producción o acumulación de ácidos. El primer caso es el más frecuente y en gran medida se explica por pérdidas gastrointestinales (diarreas) aunque la hiperalimentación, el uso de acetazolamida, otros diuréticos, fístula pancreática, ureterostomías o acidosis tubular renal proximal (tipo 2) también pueden generar este tipo de acidosis.
En el caso de hiato aniónico incrementado las causas pueden ser acidosis láctica, ce-toacidosis, ingesta de ácidos, infusiones de medicamentos, insuficiencia renal, acidosis tubular renal distal (tipo 1), acidosis tubular renal con hiporaldosteronismo hiporreninémico (tipo 4), entre otras (Cuadro 2). Existen varias nemotecnias para recordar las diversas causas (e.g. CAT MUDPILES, GOLD MARK, HARDUP) que no serán contempladas en este artículo pero que es útil conocer.1-3

El conocimiento de la fisiología ácido-base y la interpretación correcta de una gasometría arterial son dos herramientas clave para el abordaje diagnóstico de un paciente con acidosis metabólica. Es importante agregar la relevancia de la asociación de la acidosis metabólica con hipo- e hiperglicemia, cetosis y alteraciones hidroelectrolíticas.
No conocemos los detalles de la evolución clínica y de laboratorio del primer evento; sin embargo, la persistencia de acidosis metabólica de hiato aniónico incrementado, a pesar de iniciar medidas terapéuticas, asegurar la ausencia de pérdidas gastrointestinales o urinarias y descartar fuentes externas de ácidos, orientaron al clínico a considerar la acumulación de ácidos orgánicos y la posibilidad de un error innato del metabolismo.
Las enfermedades que desencadena la acumulación de ácidos orgánicos pueden ser de dos tipos:
Enfermedades adquiridas con producción endógena de ácido láctico y cetoácidos secundaria a hipoxia, diabetes descompensada y otras más.
Errores congénitos del metabolismo con acidemia orgánica como:
Las acidemias orgánicas más importantes son:
Acidemias glutáricas, propiónica, metil-malónicas o isovalérica.
Déficit múltiple de carboxilasas.
Enfermedad de orina de jarabe de arce.
Normalmente los ácidos orgánicos se meta-bolizan en productos no ácidos y se eliminan por la orina, pero en situaciones anormales se acumulan en los tejidos y líquidos del cuerpo produciendo un aumento de H+ y acidosis metabólica. Lo anterior desencadena en estas enfermedades exceso en la producción de metabolitos tóxicos con disminución en la producción y utilización de los sustratos energéticos, aumentando la producción de otros ácidos orgánicos.4,5
De acuerdo con la Guía de Referencia Rápida de Acidemias Orgánicas: acidemia metilmalónica y propiónica, del Consejo de Salubridad General, los estudios iniciales ante la sospecha de errores innatos del metabolismo deben incluir: biometría hemática completa con diferencial, examen general de orina, gasometría arterial, electrólitos séricos, glucosa, amonio en plasma, sustancias reductoras en orina, cetonas urinarias. Si hay acidosis o hipoglicemia: cuantificación de aminoácidos en plasma y orina, ácidos orgánicos en orina y lactato plasmático.6
En este caso, el perfil de los ácidos orgánicos y de los aminoácidos en orina permitió establecer el diagnóstico de acidemia metilmalónica. Esta enfermedad se debe a un grupo de errores congénitos del metabolismo de los aminoácidos ramificados, generando acumulación de ácido metilmalónico en los líquidos corporales. Esto se debe a la imposibilidad de convertir la metilmalonil-CoA en succinil-CoA en la vía del propionato, por un defecto en la proteína mutasa o en algún paso del procesamiento intracelular de cobalamina, que conduce a la síntesis de cofactor AdoCbl. Al no metabolizarse se desencadena acidosis metabólica por acumulación de cuerpos cetónicos, cetosis y ácidos orgánicos y acilcarnitinas en orina; todos específicos para cada aminoácido7 (Figura 1).
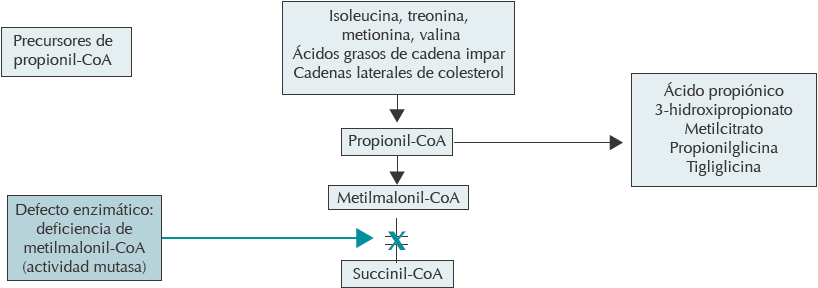
Traducido y modificado de: New England Consortium of Metabolic Programs. Disponible en: http://newenglandconsortium.org/for-professionals/acute-illness-protocols/organic-acid-disorders/methylmalonic-acidemia/
Figura 1 Defecto enzimático en acidemia metilmalónica.
Se han descrito distintos tipos de acidemia metilmalónica dando diferentes defectos bioquímicos. Aproximadamente 50% de los pacientes corresponde al grupo de mutaciones en genes que codifican la mutasa con locus en el brazo corto del cromosoma 6 divididos en grupo Mut0 (sin actividad de la mutasa y sin responder a B12) y grupo Mut- (con actividad parcial si se añade cobalamina). El resto de los pacientes presenta mutaciones en los genes requeridos para la síntesis de cobalaminas (i.e. MMAA, MMAB, MMADHC, MCEE) 7,8 (Figura 2).
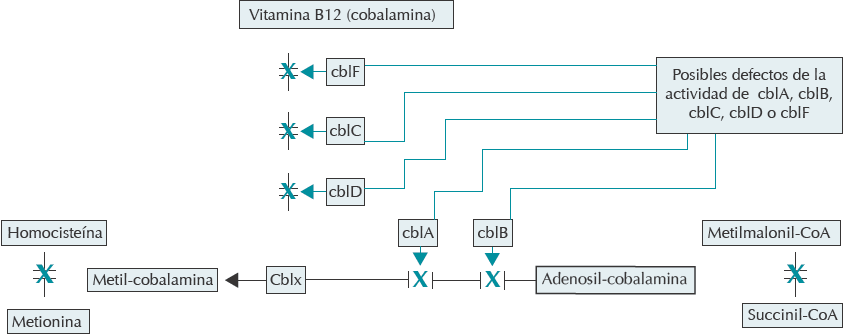
Traducido y modificado de: New England Consortium of Metabolic Programs. Disponible en: http://newenglandconsortium.org/for-professionals/acute-illness-protocols/organic-acid-disorders/methylmalonic-acidemia/
Figura 2 Posibles defectos enzimáticos de la vitamina B12 asociados con acidemia metilmalónica.
El bloqueo metabólico produce acumulación intracelular de metilmalonil-Coa y del ácido metilmalónico en el plasma y la orina. La acumulación de propionil-CoA inhibe el metabolismo intermedio de la glicina, de la piruvato deshidrogenasa y de la cetil-glutamato sintetasa. El exceso de metilmalonil-CoA y propionil-CoA compite con acetil-CoA en la síntesis de ácidos grasos, formando no fisiológicamente ácidos grasos de cadena larga impar, esto explica la hi-poglicemia, la hiperglicemia, hiperlactacidemia e hiperamonemia.7-9
La paciente, después de 14 años de ser tratada en este Instituto y con múltiples internamien-tos, falleció por descompensación grave de su patología de base asociada con un evento de abdomen agudo. Los eventos de estrés celular desencadenados por infecciones, traumatismos, cirugías o cualquier estado de catabolismo, confieren condiciones que promueven desequilibrio metabólico per se. El tratamiento dirigido a resolver el evento desencadenante siempre deberá acompañarse de un manejo metabólico intensivo y dinámico para favorecer un buen resultado. En el caso discutido la paciente presentó un deterioro catastrófico irreversible a las medidas terapéuticas proporcionadas.
HALLAZGOS ANATOMOCLÍNICOS
Se realizó estudio de autopsia a paciente con menor edad aparente a la cronológica, con cicatriz en línea media supraumbilical y gastros-tomía. Al examen macroscópico se observaban pulmones de coloración pálida alternando con zonas de aspecto congestivo (Figura 3), páncreas con áreas de aspecto congestivo alternando con áreas nodulares de color blanquecino y consistencia deleznable. El cerebro mostraba múltiples infartos cerebrales calcificados (Figura 4). La mucosa del colon presentó múltiples áreas de color blanquecino y depósito de material de aspecto fibrinoide (Figura 5).

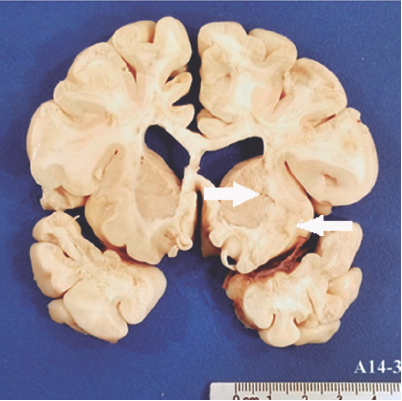
Figura 4 Corte de cerebro que muestra imágenes quísticas correspondientes a zonas de infarto y microcalcificaciones (flechas).
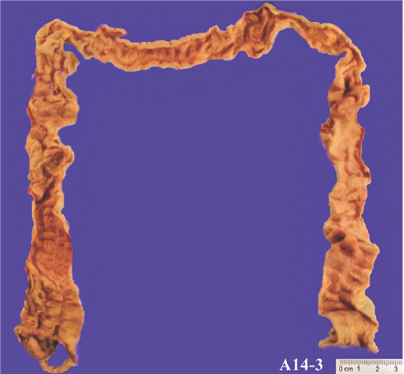
Figura 5 Imagen que muestra múltiples áreas de coloración pálida correspondientes a zonas isquémicas de la mucosa.
Dentro de los hallazgos principales en la microscopia se encontraron gastritis, ileítis y colitis isquémica focal con trombos recientes y numerosas colonias de Cándida spp (Figura 6). También se observaron pancreatitis aguda necrótica hemorrágica (Figura 7), trombosis pulmonar reciente con datos histológicos de broncoaspiración e infartos cerebrales antiguos con microcalcificaciones.
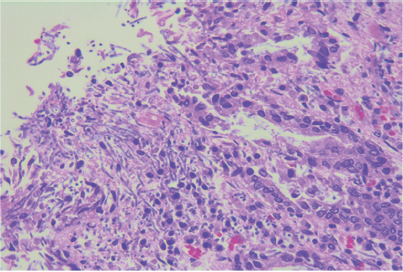
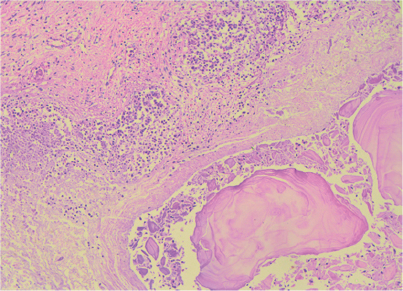
Figura 7 Fotomicrografía: ácinos pancreáticos (parte superior) parcialmente destruidos con áreas de necrosis de tejido adiposo (saponificación).
Los diagnósticos anatomopatológicos finales fueron:
Historia clínica de acidemia metilmalónica descompensada
Trombosis pulmonar reciente con datos histológicos de broncoaspiración
Hemofagocitosis en médula ósea
Datos histológicos de choque:
Infartos cerebrales antiguos calcificados
Laringitis aguda leve
Cistitis crónica leve hemorrágica
Cistoadenomas serosos bilaterales de ovario
Estatus posgastrostomía
DISCUSIÓN
Es importante resaltar que si bien esta paciente no cuenta con un diagnóstico molecular el hecho de que haya respondido al inicio del tratamiento incluyendo vitamina B12, y lograra cierta estabilidad, hace pensar que pertenece a cualquiera de los grupos cblA, mut- o cblB. En la literatura especializada se describe que estos pacientes suelen tener mejor pronóstico en general, aunque las alteraciones neurológicas suelen desarrollarse en grado variable. A pesar de 14 años de seguimiento una descompensación metabólica asociada con un evento de abdomen agudo terminó con la vida de la paciente. Los lineamientos para el tratamiento de las crisis metabólicas están descritos en varios documentos y los pediatras debemos estar familiarizados con ellos10-14 (Cuadro 3).
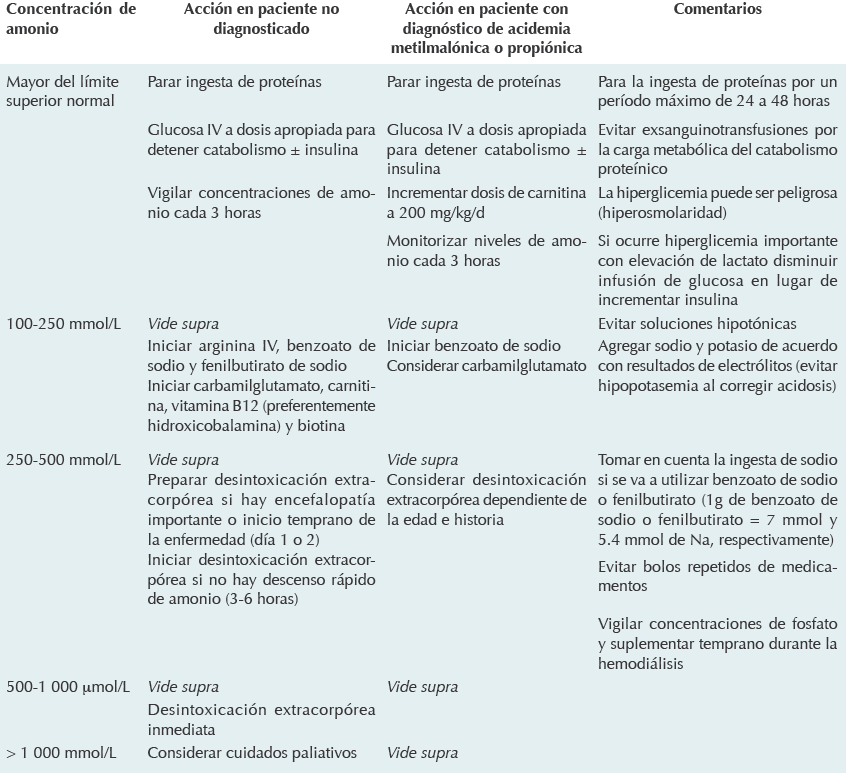
Traducido y modificado de: Baumgartner et al, Orphanet Journal of Rare Diseases 2014;9:130. Disponible en: http://www.ojrd.com/content/9/1/130
Cuadro 3 Manejo de la crisis metabólica
Las crisis metabólicas frecuentemente se acompañan de dolor abdominal, vómito y ataque al estado general en grados variables. Estos síntomas también son comunes en la pancreatitis. Si el clínico no considera esta posibilidad el tratamiento dirigido a resolver la crisis metabólica no será suficiente. Otros autores han descrito la incidencia de pancreatitis aguda y crónica en pacientes con acidemias orgánicas. Kahler y sus colaboradores reportaron la casuística de cinco instituciones pediátricas de tercer nivel de atención de pacientes con acidemias orgánicas de aminoácidos de cadena ramificada. Ellos juntaron la información de 108 niños y describieron 7 pacientes con pancreatitis aguda y 2 con pancreatitis crónica (5 con acidemia metilmalónica, 3 con acidemia isovalérica y 1 con enfermedad de orina de jarabe de arce) con 3 fallecimientos. Cabe resaltar que en una encuesta realizada en paralelo al mismo estudio se identificaron otros 60 casos pediátricos de pancreatitis en los que se diagnosticaron 3 pacientes con acidemias orgánicas.14,15
Marquard y sus colaboradores describieron otro caso de un niño de 5 años con acidemia metilmalónica que desarrolló pancreatitis aguda en 5 ocasiones, posteriormente desarrolló pancreatitis crónica. En ese artículo reunió la información de otros 13 pacientes que en total cursaron con 30 casos de pancreatitis y hubo 4 fallecimientos. Ambas publicaciones coinciden en que la pancreatitis es una complicación que ocurre más frecuentemente en estos pacientes que en la población general y con una mayor tasa de complicaciones y mortalidad; es por ello que ante un cuadro mínimamente sugestivo se deberá descartar (i.e. todas las crisis metabólicas que se presenten con vómito, hiporexia y ataque al estado general).16
El hecho de que clínica e histológicamente la paciente haya cursado con una pancreatitis aguda necroticohemorrágica con niveles de amilasa y lipasa normales no es lo más común y puede desorientar al pediatra. Ya se han descrito diferentes series de casos con un comportamiento similar: al menos 7 casos se han reportado con pancreatitis aguda en los que la presentación clínica era concordante con dicho proceso pero los niveles de amilasa y lipasa estuvieron en rangos normales a lo largo de sus evoluciones, identificando los procesos inflamatorios por imagen (ultrasonografía, tomografía, resonancia magnética) o biopsia post mortem.17-21 Los autores discuten que la determinación de las concentraciones de amilasa y lipasa pueden verse influenciadas por la presencia de un suero lipémico (con hipertrigliceridemia o hipercoles-terolemia), por ser agudizaciones de un proceso crónico preexistente o bien por ser eventos agudos graves con afección total del páncreas (que pueden cursar con niveles de amilasa y lipasa normales) una vez que el tejido pancreático se consumió. También la gravedad y el momento en que se solicitan los estudios pueden modificar el comportamiento de las concentraciones de amilasa y lipasa.17-21
Con excepción de los infartos cerebrales antiguos calcificados, que pudieron ser resultado de los diferentes eventos previos con los que cursó la paciente, la mayoría de los hallazgos histopatológicos descritos son compatibles con un evento agudo catastrófico. La evidencia de afección multisistémica de inflamación, hipoxia, isquemia y coagulación intravascular diseminada son congruentes con la nula respuesta al tratamiento establecido y el desenlace de la paciente.
Los errores innatos de metabolismo son un reto para los pacientes, sus familias y para los médicos que las atendemos. Un enfoque multi-disciplinario y la experiencia acumulada de una institución como la nuestra permiten mejorar las condiciones generales de estos pacientes en la mayoría de los casos. El análisis de este caso nos ha permitido compartir los distintos escenarios que ocurren en un mismo paciente con acidemia metilmalónica a lo largo de su vida y resaltar la importancia de los datos clínicos sobre las alteraciones bioquímicas.

