










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa pediátrica de México
versión On-line ISSN 2395-8235versión impresa ISSN 0186-2391
Acta pediatr. Méx vol.35 no.6 México nov./dic. 2014
Caso de sesión anatomo-clínica
Diarrea crónica intratable asociada con diarrea fenotípica de la infancia (síndrome tricohepatoentérico)
Intractable chronic diarrhea associated with phenotypic diarrhea of infancy (tricho-hepato-enteric syndrome)
José Francisco Cadena-León1 y Rodolfo Rodríguez-Jurado2
1 Departamento de Gastroenterología y Nutrición Pediátrica.
2 Departamento de Patología. Instituto Nacional de Pediatría, México, D.F.
Correspondencia:
Dr. Rodolfo Rodríguez Jurado
Departamento de Anatomía Patológica
Instituto Nacional de Pediatría
Insurgentes Sur 3700-C
CP. 04530 México, D.F.
Tel.: 10840900 ext. 1463
Recibido: 10 de septiembre 2014.
Aceptado: 1 de octubre 2014.
INTRODUCCIÓN
La diarrea crónica intratable de la infancia se caracteriza por evacuaciones disminuidas de consistencia, muy frecuentes, con duración de más de cuatro semanas, acompañadas de malabsorción intestinal, desnutrición grave desde el nacimiento hasta los tres meses de vida y dependiente de nutrición parenteral.1 La diarrea fenotípica de la infancia o síndrome tricohepatoentérico es una entidad relativamente nueva, descrita en 1994, con una incidencia estimada de 1 por cada millón de nacidos vivos. Tiene patrón de herencia autosómico recesivo asociado con mutaciones en dos genes: SKIV2L y TTC37, que son cofactores que forman el complejo humano ski. La forma de presentación clásica se caracteriza por diarrea crónica intratable, dismorfias faciales, anormalidades en el pelo, inmunodeficiencia y retraso del crecimiento intrauterino; tiene mortalidad de 60% en el primer año de vida. Este artículo describe el caso de un lactante con diarrea crónica intratable cuyo estudio post mórtem permitió excluir algunas entidades nosológicas y proporcionó evidencia para diagnóstico probable de diarrea fenotípica de la infancia o síndrome tricohepatoentérico.
RESUMEN CLÍNICO
Lactante femenino de 15 meses de edad, producto de segunda gesta, con embarazo de evolución normal complementado con ácido fólico y fumarato ferroso. Hija de mujer de 22 años de edad referida como sana y sin antecedentes relevantes. Un hermano de 4 años de edad también referido como sano. Nació por vía abdominal (por cesárea previa) a las 39 semanas de gestación con peso de 3 150 g (centil 42.8) y talla de 47 cm (centil 12.4), perímetro craneal de 32 cm (centil 3), Apgar 8/9. Sufrió fractura de húmero izquierdo durante el nacimiento que fue tratada con inmovilización. Fue alimentada con leche humana, fórmula de inicio, agua de arroz endulzada con piloncillo y anís desde el primer mes de vida.
Su padecimiento inició a los 14 días de vida con 10 evacuaciones líquidas, sin sangre; fiebre no cuantificada, somnolencia y disnea. Fue traída al servicio de urgencias de nuestro hospital por deshidratación grave. Fue tratada con dos cargas de cristaloides a 10 mL/kg/do, bicarbonato de sodio y bicarbonato de potasio con lo que la paciente mostró mejoría. Se agregaron antibióticos por sospecha de sepsis neonatal temprana. Se documentó acidosis metabólica hiperclorémica de hiato aniónico normal.
En la exploración física se documentó hipertelorismo, puente nasal ancho, epicanto bilateral, paladar alto, baja implantación de pabellones auriculares y pliegue simiesco incompleto.
Se solicitaron estudios para descartar errores innatos del metabolismo: ácidos orgánicos en orina por cromatografía de gases, perfil de aminoácidos, acilcarnitinas, espectrometría de masa en tándem y tamiz metabólico ampliado, que fueron reportados dentro de los parámetros normales; fue egresada para continuar seguimiento ambulatorio. A los dos meses de edad regresó por tener evacuaciones líquidas (de tres a diez veces por día) que provocaban deshidratación grave. A su ingreso pesó 3.5 kg y midió 50 cm, talla/edad (TE) 89.3%, peso/edad (PE) 76%; se documentó acidosis metabólica. Ante el cuadro de diarrea crónica y desnutrición se le administró fórmula extensamente hidrolizada de proteínas de suero a 15%. Se documentó, además, dermatosis localizada en cabeza y piel cabelluda, en regiones frontal y biparietal considerada como dermatitis seborreica, así como dermatitis de la zona del pañal con sobreinfección por Candida que recibió tratamiento con itraconazol. Durante la hospitalización desarrolló infección de vías urinarias por hongos tratada con fluconazol e infecciones asociadas con catéter, en múltiples ocasiones, tratadas con ceftriaxona y vancomicina.
Se realizaron estudios de videopanendoscopia y rectosigmoidoscopia. Macroscópicamente la mucosa esofágica no mostró alteraciones; el estómago tenía la mucosa eritematosa; se observó bulbitis nodular y en rectosigmoides no se observaron alteraciones. Las biopsias tomadas a los 3 y a los 11 meses de vida mostraron atrofia moderada focal en el duodeno con infiltrado inflamatorio en lámina propia en los que destacó la presencia de hasta 15 eosinófilos por campo a seco fuerte. En el colon se encontraron más de 20 eosinófilos por campo de alto poder (Figuras 1, 2, 3 y 4). En su momento estos hallazgos se consideraron como parte de una enteropatía eosinofílica y se dio tratamiento con fórmula elemental, nutrición parenteral total y esteroides sistémicos. Durante su evolución se cambió la dieta a fórmula de pollo (artesanal) con la que mostró mejor tolerancia. Se suspendió la nutrición parenteral total a los 11 meses.




A los 11 meses de hospitalización y 13 meses de edad pesó 4 970 g, midió 63 cm, peso para la talla (P/T) 75.3%, talla para la edad (T/E) 83.77%; peso para la edad (P/E) 54%. Se le diagnosticó desnutrición grave.
Se sospechó una probable inmunodeficiencia primaria (alteración en la inmunorregulación) por las infecciones recurrentes, diarrea crónica y desnutrición grave, por lo que se solicitaron inmunoglobulinas, complemento, citometría de flujo y reducción de nitroazul de tetrazolio que no mostraron alteraciones. Se inició tratamiento con ciclosporina a 3 mg/kg/d, metilprednisolona y gammaglobulina intravenosa 2 g/kg/do en dos ocasiones con mejoría clínica.
A los 15 meses tuvo una infección por el uso de catéter, con datos de respuesta inflamatoria sistémica. Se le trató con vancomicina, meropenem y anfotericina B; no hubo mejoría y tuvo paro cardiorrespiratorio.
HALLAZGOS ANATOMOPATOLÓGICOS
Además del hipertelorismo y el puente nasal ancho referidos, llamó la atención una hipertricosis facial peculiar en la zona por arriba del labio superior y el surco labio mentoniano (Figura 5). El pelo era lacio.

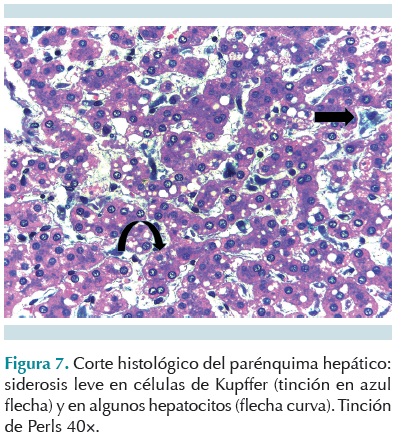
El estudio morfológico del tubo digestivo mostró atrofias leves y focales en el intestino delgado y el colon, con hiperplasia de células caliciformes sin evidencia de infiltrado inflamatorio eosinofílico. En el sistema inmunológico se encontró atrofia tímica moderada con corpúsculos de Hassall (Figura 6); disminución de la población linfoide en los ganglios linfáticos y el bazo. Hubo hemosiderosis hepática leve (Figura 7) y datos anatómicos de septicemia con bronconeumonía incipiente, hepatitis y esplenitis sépticas. En los cortes histológicos de riñón se encontró nefrocalcinosis (Figura 8).


DISCUSIÓN
En esta paciente con diarrea crónica recurrente e intratable, de inicio en la segunda semana de vida, los estudios de biopsia con microscopia electrónica permitieron descartar enfermedad de inclusión microvellosa (Figura 4). Además, se descartó enteropatía en penacho (displasia intestinal) en las biopsias de intestino delgado ya que no existía atrofia vellosa grave con formación de "mechones" de epitelio intestinal regenerativo ni enterocitos individuales en forma de "lágrima". Otra entidad que se descartó con las biopsias de intestino delgado fue la enteropatía autoinmunitaria, ya que no había atrofia total de vellosidades con destrucción de criptas y apoptosis en la base de las criptas.1
Los hallazgos morfológicos de las biopsias intestinales, las dismorfias faciales (hipertelorismo y puente nasal ancho con pelo escaso) (Figura 5), aunados a la sospecha de inmunodeficiencia primaria (alteración en la inmunorregulación), apoyó las probabilidades de diarrea fenotípica de la infancia o síndrome tricohepatoentérico. Esta entidad fue descrita en 1994 por Girault y sus colaboradores como diarrea intratable fenotípica o sindromática, asociada con dismorfismo facial, retraso en el crecimiento intrauterino, inmunodeficiencia y anormalidades en el pelo.2 La diarrea sindrómica/síndrome tricohepatoentérico es la primera afección mendeliana asociada con alteración en los exosomas citosólicos de las células. Verloes y sus colegas describieron la diarrea fenotípica de la infancia o síndrome tricohepatoentérico como diarrea intratable con fenotipo de hemocromatosis neonatal y anormalidades en el pelo.3 Se han agrupado en un solo síndrome porque las características clínicas principales son idénticas. Varios autores han identificado mutaciones en el gen TTC37 en 21 individuos con el análisis de mapeo homocigoto después de realizar arreglo de polimorfismo de nucleótido único (SNP por sus siglas en inglés); dicho gen fue identificado en el locus 5q14.3-5q21.2 y codifica una proteína repetida, tetratricopeptídica no caracterizada llamada thespin (proteína del síndrome tricohepatoentérico por sus siglas en inglés). El efecto de dicha mutación se piensa que es multisistémico aunque se han encontrado, con estudios de inmunofluorescencia confocal, inadecuada distribución o expresión disminuida de múltiples proteínas transportadoras apicales en enterocitos yeyunales.4
Fabre y su grupo observaron que 6 de 15 individuos que mostraban características clínicas de diarrea fenotípica de la infancia o síndrome tricohepatoentérico no mostraron la mutación en TTC37; consideraron a TTC37 homólogo al gen SKI3 de levaduras y este es un componente clave de un complejo multiproteínico denominado Ski, requerido para la vigilancia exosomal de descomposición del ARN y la vigilancia de la expresión de la fidelidad genética del propio ARN. Buscaron mutaciones en el gen ortólogo humano de SKI3 que resultó en la identificación de mutaciones deletéreas en el gen SKIV2L en los seis individuos referidos.5 En el Cuadro 1 se resumen los hallazgos de estos autores con los datos observados en nuestra paciente. Ellos mismos señalan otro gen candidato para los casos potenciales donde la pesquisa de mutaciones conocidas sea negativa: el gen WDR61, ortólogo humano del tercer cofactor SKI8.
La dermatitis seborreica que junto con diarrea crónica, falla de medro e infecciones recurrentes, hizo pensar en descartar fenotipo de Leiner, aunque el complemento fue normal y no se corroboró un defecto inmunitario específico. Cabe mencionar que tampoco se documentó xerosis ni las manchas "café con leche" que se han descrito en los pacientes con diarrea fenotípica de la infancia o síndrome tricohepatoentérico.
En el aspecto inmunológico de diarrea fenotípica de la infancia o síndrome tricohepatoentérico, originalmente se describió inmunodeficiencia funcional de células T6 con pruebas de piel antígeno específicas defectuosas y producción defectuosa de anticuerpos, a pesar de poder cursar con niveles séricos de inmunoglobulinas normales. En informes adicionales con pruebas inmunológicas más extensas se ha encontrado hiper-IgA monoclonal con hipo-IgG. Nuestra paciente cursó con intertrigo por Candida, infección de vías urinarias sin germen aislado y murió por complicaciones infecciosas (i.e. sepsis). Es muy probable que un estudio más detallado de la función inmunológica hubiera mostrado la disfunción. Otro hallazgo que no se ha descrito en la literatura es la acidosis metabólica hiperclorémica con hiato aniónico normal y nefrocalcinosis que obliga a considerar el diagnóstico de acidosis tubular renal; en este caso no se asoció con síndrome de Fanconi (glucosuria, aminoaciduria o fosfaturia) pero puede cursar con diarrea crónica y detención de crecimiento.
El tratamiento de la diarrea fenotípica de la infancia o síndrome tricohepatoentérico consiste en mantener adecuados crecimiento y desarrollo, y en evitar complicaciones asociadas como desnutrición grave y sepsis mediante el empleo de fórmulas especiales, nutrición parenteral e inmunoglobulina G intravenosa.
Cabe destacar, desde el punto de vista clínico, que a pesar de haber recibido tratamiento médico vigoroso, abarcando aspectos nutricionales, infecciosos e inmunológicos, la paciente no tuvo la respuesta esperada. Sólo hubieron mejorías parciales y recaídas constantes del síndrome diarreico, que no permitieron recuperar el estado de salud de la paciente; los hallazgos histológicos en el estudio post mórtem no documentaron actividad inflamatoria ni eosinofílica en el intestino.
En el Cuadro 1 se muestran los hallazgos más frecuentemente observados en 24 pacientes reportados en diversos estudios. De los diez hallazgos más frecuentes siete se observaron en nuestra paciente. La probabilidad de encontrar alguna de las mutaciones descritas es alta, para corroborar el diagnóstico en forma contundente. Asimismo, el ejercicio clinicopatológico de excluir otras posibilidades, como queda demostrado en este artículo, aunque sea un diagnóstico no corroborado, permite ampliar nuestro panorama diagnóstico. Un caso nuevo potencial de la consulta de gastroenterología pediátrica ha iniciado su estudio diagnóstico, lo que sin duda fue motivado por el estudio de autopsia de esta paciente y su discusión.
REFERENCIAS
1. Russo P. Enteropathies associated with chronic diarrea and malabsorption. En: Russo P, Ruchelli E, Piccoli D (eds). Pathology of pediatric gastrointestinal and liver disease. Capítulo 4. New York: Springer, 2004; pp. 63-99. [ Links ]
2. Girault D, Goulet O, Le Deist F, Brousse N, Colomb V, Césarini J, et al. Intractable infant diarrhea associated with phenotypic abnormalities and immunodeficiency. J Pediatr 1994;125:36-42. [ Links ]
3. Verloes A, Lombet J, Lambert Y, Hubert A, Deprez M, Fridman V, et al. Tricho-hepato-enteric syndrome: Further delineation of a distinct syndrome with neonatal hemo-chromatosis phenotype, intractable diarrhea, and hair anomalies. Am J Med Genet 1997;68:391-395. [ Links ]
4. Hartley J, Zachos N, Dawood B, Donowitz M, Forman J, Pollett R, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (Phenotypic Diarrhoea of infancy). Gastroenterology 2010;138(7):2388-2398. [ Links ]
5. Fabre A, Charroux B, Martinez-Vinson C, Rochelaure B, Odul E, Sayar E, et al. SKIV2L mutations cause syndromic diarrea, or tricohepatoenteric syndrome. Am J Hum Genet 2012;90(6):689-692. [ Links ]
6. Goulet O, Vinson C, Roquelaure B, Brouse N, Bodemer C, Cézard J. Syndromic (phenotypic) diarrhea in early infancy. Orphanet J Rare D 2008;3:1-6. [ Links ]
Nota
Este artículo debe citarse como
Cadena-León JF, Rodríguez-Jurado R. Diarrea crónica intratable asociada con diarrea fenotípica de la infancia (síndrome tricohepatoentérico). Acta Pediat Mex 2014;35:483-489.

