










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa pediátrica de México
versión On-line ISSN 2395-8235versión impresa ISSN 0186-2391
Acta pediatr. Méx vol.35 no.4 México jul./ago. 2014
Caso clínico de interés especial
Tumoraciones intracardiacas múltiples en un neonato. Reporte de caso
Multiple intracardiac tumors in a neonate. A case report
Ben David Valdés-Ramos1, Fabiola Pérez-Juárez2, Carlos López-Candiani3
1 Residente de Neonatología.
2 Cardiólogo Pediatra adscrito al Laboratorio de Ecocardiografía.
3 Neonatólogo, M en C. Jefe del Departamento de Neonatología. Instituto Nacional de Pediatría, México. D. F.
Correspondencia:
Dr. Carlos López-Candiani
Departamento de Neonatología
Instituto Nacional de Pediatría
Insurgentes Sur 3700 C
CP. 04530, México, D.F.
Tel: 10 84 09 00 ext. 1352
mensaje55@hotmail.com
Recibido: 9 de octubre 2013
Aceptado: 27 de mayo 2014
RESUMEN
Los tumores intracardiacos son poco frecuentes en niños y más aún en los neonatos. La mayoría de este tipo de lesiones son rabdomiomas; usualmente son múltiples y de diferentes tamaños, de localización ventricular y muy rara vez se encuentran en las aurículas. Los rabdomiomas se pueden asociar con esclerosis tuberosa de 56 a 86% de los casos.
Actualmente, gracias a las consultas prenatales, las tumoraciones fetales intracardiacas pueden ser diagnosticadas desde el segundo trimestre de la gestación. La gravedad dependerá de la localización del tumor y del efecto hemodinámico que ejerza, más que del tamaño. El seguimiento con ultrasonidos prenatales seriados revela la velocidad del crecimiento tumoral. Se piensa que debido a una estimulación hormonal materna intraútero, entre el segundo y tercer trimestres presenta una tasa de crecimiento acelerada que disminuye a partir de la semana 35.
Se describe el caso de una paciente con diagnóstico prenatal de tumores intracardiacos, referida al departamento de neonatología de un hospital público de especialidades pediátricas.
Palabras clave: tumor cardiaco neonatal, rabdomioma, esclerosis tuberosa, ecocardiografía.
ABSTRACT
Intracardiac tumors are uncommon in children, and to a lesser extent in newborns. Most of these tumors are rabdomiomas, which usually vary in size and number. The rabdomioma is located most of the times in the ventricles, and rarely in the atrium. The rabdomiomas can be associated with tuberous sclerosis from 56% to 86% of the cases.
Nowadays, intracardiac fetal tumors can be detected through prenatal ultrasound, after the second gestation trimester. The tumor location and the hemodynamic effects will be the most important aspects that determine the severity.
Serial prenatal ultrasound monitoring reveals the tumoral growth speed. It's believed that, due to an in utero maternal stimulation, an accelerated growth rate is presented during the second and third trimester, and it will slow down after the 35th week.
This is a case-report of a patient with the prenatal diagnosis of an intracardiac mass, referred to the neonatology department of the Instituto Nacional de Pediatría.
Key words: Cardiac tumor, fetal rhabdomyoma, sonography, tuberous sclerosis.
ANTECEDENTES
Los tumores cardiacos en el grupo etario de 0 a 18 años son extremadamente raros, más aún en neonatos. Estos tumores son considerados benignos por sus características morfológicas, suelen presentarse con una amplia variedad de manifestaciones clínicas, por lo cual, rara vez se detectan en la etapa neonatal y representan un reto diagnóstico y terapéutico para el clínico.1
Distintas publicaciones de tumoraciones cardiacas primarias informan una incidencia de entre 0.08 y 0.28% por cada 1 000 recién nacidos vivos, con una distribución mundial similar entre niños y niñas. El más común en la edad pediátrica es el rabdomioma (60%) seguido por el teratoma (25%) y, en tercer lugar, el fibroma (12%). Los rabdomiomas usualmente son múltiples y de diversos tamaños. Su localización más común es en sitios múltiples (43%), en los ventrículos exclusivamente (41%), en el septo interventricular (15%) y muy rara vez en las aurículas.2
De los tumores cardiacos primarios 90% es histológicamente benigno. Los rabdomiomas son considerados de mal pronóstico cuando causan obstrucción a los flujos de entrada o de salida del corazón, regurgitación del aparato valvular, se acompañan de arritmias, derrame pericárdico, insuficiencia cardiaca o feto hidrópico.3 Los rabdomiomas se caracterizan, desde el punto de vista ultrasonográfico, por ser tumoraciones redondeadas, homogéneas, hiperecogénicas, usualmente múltiples y de localización predominantemente ventricular; generalmente guardan relación con el septo interventricular.4
Existe una incidencia de entre 9 y 18% de síndrome de Wolff-Parkinson-White en los pacientes con rabdomioma y puede manifestarse por muerte súbita o arritmia supraventricular. También se puede asociar con esclerosis tuberosa entre 50 y 64% de los casos,5 enfermedad autosómica dominante caracterizada por retraso mental, convulsiones y lesiones cutáneas.6 Estos tumores suelen tener involución espontánea completa antes de los 4 años de edad. A continuación se describe el caso de un neonato con múltiples tumoraciones intracardiacas, tratado en el departamento de neonatología del Instituto Nacional de Pediatría.
CASO
Neonato hijo de madre de 38 años y padre de 42 años de edad, sanos, con 2 hermanos vivos y sanos de 7 y 5 años. La madre llevó un control prenatal regular a partir del primer mes de gestación; fue suplementada con hierro y ácido fólico a partir de la octava semana. Durante el embarazo cursó con cervicovaginitis en el primer trimestre, no tratada. A las 34 semanas, en un ultrasonido obstétrico se observaron masas intracardiacas, por lo que fue referida a un hospital regional y, de ahí, a un centro de referencia en donde se le programó para cesárea a la semana 39. Se obtuvo un producto femenino, con peso al nacer de 2 325 g, talla de 48 cm, Apgar 8/9, Silverman y Andersen de 2, placenta y el líquido amniótico sin alteraciones.
A la exploración física, al nacimiento, se encontraron como datos anormales apéndices auriculares bilaterales; soplo sistólico grado II/VI, en cuarto espacio intercostal línea medio clavicular; a nivel de tronco una dermatosis localizada que afectaba la región posterosuperior paravertebral izquierda, caracterizada por 3 máculas hipopigmentadas, la menor de 3 mm de diámetro y 2 más de 8 mm de diámetro, de limites bien definidos, lanceoladas.
Como parte de su abordaje diagnóstico se le practicó electrocardiograma que evidenció ritmo sinusal y predominio de fuerzas derechas. (Figura 1) Además, se le tomó una radiografía de tórax donde se encontró un índice cardiotorácico de 0.62 a expensas de las cavidades derechas. (Figura 2)

El ecocardiograma reportó anatomía segmentaria normal. Foramen oval permeable de 2 mm con cortocircuito restrictivo de izquierda a derecha, con gradiente instantáneo máximo de 18 mmHg y gradiente medio de 9 mmHg. Se observaron tumoraciones intracardiacas múltiples sugestivas de rabdomiomas. La mayor de ellas, en la aurícula izquierda, redondeada, de 15 × 12 mm, muy cercana a la válvula mitral. Cabe destacar que las masas intraauriculares se encuentran en menos de 1% de los casos reportados en la literatura especializada. Se encontró otra tumoración en el ventrículo derecho, más pequeña (10 × 8 mm), de aspecto bilobulado, dependiente del septo interventricular; dos más en el ventrículo izquierdo, una en el tercio medio del septo interventricular, de 4 × 4 mm, redondeada, y otra de 6 × 3 mm en la base del músculo papilar. (Figuras 3, 4 y 5)



Además, se observó insuficiencia mitral moderada, condicionada por impedimento en el cierre de la valva anterior y secundario a la tumoración en la aurícula izquierda. Insuficiencia tricuspídea trivial con gradiente instantáneo máximo de 70 mmHg. Sin datos de obstrucción al llenado ventricular izquierdo ni al tracto de salida del ventrículo derecho. Persistencia del conducto arterioso de 2 mm de boca pulmonar, 9 mm de longitud y 2 mm de boca aórtica, con cortocircuito bidireccional pero franco predominio de derecha a izquierda. Hipertensión arterial pulmonar suprasistémica. Cualitativamente, buena función biventricular.
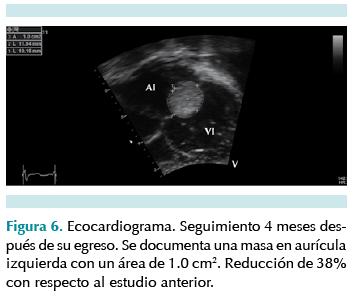
Durante su estancia la paciente presentó una evolución clínica favorable, fue alimentada de forma habitual con seno materno; no se documentaron episodios de crisis convulsivas. Recibió tratamiento médico con captopril (0.025 mg/kg 3 veces al día), furosemida (0.5 mg/kg 3 veces al día) y espironolactona (3 mg/kg/día). Fue evaluada por genética, oftalmología, dermatología y neurología. Se estableció el diagnóstico de esclerosis tuberosa de acuerdo con los criterios establecidos en la Conferencia de Consenso sobre el Complejo de Esclerosis Tuberosa de 2012. En sesión medicoquirúrgica se concluyó que, dado que estos tumores suelen tener involución espontánea completa antes de los 4 años de edad y que al momento no presentaba repercusión hemodinámica, valorando el riesgo-beneficio, la conducta fuera dar seguimiento estrecho por consulta externa y realizar alguna intervención si hubiese necesidad de ello. Luego de constatar buena ganancia ponderal y sin otra manifestación clínica de su enfermedad egresó para seguimiento por consulta externa. Cuatro meses después del egreso, con un nuevo ecocardiograma, se demostró una reducción del rabdomioma localizado en la aurícula derecha de 0.6 cm2, lo cual representaba un 38% de su tamaño original, sin condicionar repercusión hemodinámica (Figura 6) y con disminución del gradiente a través de la válvula mitral de 3.4 mmHg de gradiente medio; además de tomografía de cráneo y ultrasonido abdominal normales.
DISCUSIÓN
Los rabdomiomas son el tumor intracardiaco más común en el periodo neonatal, y mantienen una relación muy estrecha con la esclerosis tuberosa. Frecuentemente se presenta como múltiples tumoraciones con tendencia a la regresión con el paso del tiempo. Existe riesgo de obstrucción del flujo sanguíneo, embolia, disfunción valvular, disfunción ventricular, arritmias e isquemia miocárdica por compresión de las coronarias.
La ecografía fetal provee información importante sobre la anatomía, función y ritmo cardiacos. Ciertas condiciones son indicativas para la realización de un ecocardiograma fetal, como son: exposición a teratógenos (isotretinoína, fenitoína, ácido valproico, carbamacepina, litio) o bien historia familiar de defectos cardiacos o síndromes mendelianos como esclerosis tuberosa, Marfan, Ellis-Van Creveld, Noonan, DiGeorge.7 La detección prenatal de tumoraciones intracardiacas ha permitido la planeación de los nacimientos de pacientes con estos hallazgos, en centros especializados, en donde se pueden atender las complicaciones y dar el consejo genético apropiado.8
Los nevos hipopigmentados pueden ser la manifestación cutánea más temprana de la esclerosis tuberosa.9 Aunque nevos hipopigmentados aislados pueden ser una variante normal en los neonatos, la presencia de 3 manchas o más, independientemente de su localización, debe alertarnos para buscar signos o síntomas, historia familiar para descartar el complejo de esclerosis tuberosa. Estas máculas hipopigmentadas pueden ser uno de los indicadores clínicos más tempranos para este diagnóstico porque ocurren en aproximadamente 80% de los casos confirmados y, generalmente, están presentes al nacimiento.10
Las manifestaciones de la esclerosis tuberosa, incluyendo crisis convulsivas, tumor subependimario de células gigantes, insuficiencia renal y linfangioleiomiomatosis, pueden contribuir a la morbilidad y a la mortalidad de estos pacientes. Con el fin de hacer un diagnóstico temprano y ofrecer un tratamiento oportuno a las posibles complicaciones de la esclerosis tuberosa, en 2012, en la Conferencia de Consenso de los Institutos Nacionales de Salud sobre el Complejo de Esclerosis Tuberosa, quedó establecido el protocolo de diagnóstico y vigilancia para la esclerosis tuberosa.13 (Cuadro 1) La información resumida en el Cuadro 1 puede resultar de utilidad para que los médicos que no están en contacto frecuente con esta enfermedad reconozcan las recomendaciones de los expertos y puedan llevarlas a cabo en su práctica cotidiana.
Las tumoraciones intracardiacas, reveladas por ultrasonido prenatal, permitieron un elevado índice de sospecha de esclerosis tuberosa. Aunque la mayoría de las masas cardiacas fueron asintomáticas, el alto índice de sospecha guió a una evaluación dirigida y permitió el diagnóstico temprano de la enfermedad en la primera semana de vida. Cabe mencionar que el hallazgo de un rabdomioma intraauricular es excepcional; la gran mayoría de los rabdomiomas son de localización ventricular y en este caso, de los múltiples rabdomiomas, el de mayor tamaño se encontró en la aurícula izquierda y afectaba negativamente el funcionamiento del aparato valvular mitral. Sin embargo, gracias a un estrecho seguimiento pudo corroborarse la involución de la masa sin llegar a generar inestabilidad hemodinámica, ni efectos deletéreos sobre la paciente.
Los pacientes con esclerosis tuberosa en los que la alteración genética corresponde a una mutación que inactiva el gen TSC2 (cromosoma 16p13, que codifica para la proteína tuberina) se han asociado en mayor número y con una mayor severidad en las manifestaciones clínicas (incluyendo los rabdomiomas cardiacos que aparecen en 12% como signo de presentación) comparándolos contra los que tienen una alteración del gen TSC1 (cromosoma 9p34, que codifica para la proteína hamartina) o contra los que no se identifica mutación alguna.11 La mayoría de los pacientes con esclerosis tuberosa y tumoraciones intracardiacas son menores de 2 años de edad, probablemente debido a la tendencia a la regresión de estos tumores conforme crecen los pacientes. Sin embargo, se ha documentado el crecimiento o aparición, de novo, de rabdomiomas intracardiacos en adolescentes.
Estudios recientes revelan que la inhibición de la proteína mTOR contribuye a la regulación de síntesis proteínica, metabolismo, diferenciación, crecimiento y migración celular, elementos que se ven perdidos en la esclerosis tuberosa por deficiencia del complejo tuberina/hamartina. De forma incidental se descubrió, en el seguimiento ecocardiográfico de un paciente con esclerosis tuberosa, con rabdomiomas intracardiacos, que luego de la administración de everolimús (un inhibidor de mTOR) como manejo para astrocitoma subependimario de células gigantes, resolución casi completa del rabdomioma que había sido detectado de forma prenatal y se había mantenido en vigilancia, y sin cambios, desde su nacimiento hasta los 5 años de edad.12
CONCLUSIONES
Presentamos el caso de un feto diagnosticado, a las 34 semanas de gestación por ultrasonido, con rabdomiomas en la aurícula izquierda y en ambos ventrículos. Gracias a la coordinación interinstitucional se planeó el nacimiento del producto para que, desde su nacimiento, recibiera la atención médica multidisciplinaria que involucró neonatólogos, cardiólogos, genetistas, oftalmólogos, neurólogos, estomatólogos y dermatólogos. El trabajo conjunto contribuyó para establecer el diagnóstico oportuno y se pudo ofrecer el tratamiento médico que ha permitido la evolución favorable de la paciente. Se mantiene seguimiento a través de la Consulta Externa del Instituto Nacional de Pediatría ya que, si bien los síntomas de insuficiencia cardiaca congestiva mejoraron debido a la involución natural de los rabdomiomas, la esclerosis tuberosa es una enfermedad sistémica que ocasiona diversos síntomas en múltiples órganos y sistemas; por tanto, una detección temprana de las probables complicaciones puede mejorar su supervivencia y su calidad de vida.
REFERENCIAS
1. Cigarroa JA, García Y, Gutiérrez LY, Jiménez S, Martínez A, Ortegón J, et al. Rabdomioma cardiaco tratado quirúrgicamente con éxito y revisión de la literatura. Rev Arch Cardiol Méx 2005;75:S113-S17. [ Links ]
2. López N, Rodríguez G, Vegas G, De La Calle M, González A. Tumores cardiacos fetales: diagnóstico ecográfico, evolución y tratamiento. Rev Chil Obstet Ginecol 2011;76(3):147-154. [ Links ]
3. Isaacs H. Fetal and Neonatal Cardiac Tumors. Pediatric Cardiology 2004;25:252-73. [ Links ]
4. Chao A, Chao, A, Wang T, Chang Y, Chang Y, Hsieh C, et al. Outcome of antenatally diagnosed cardiac rhabdomyoma: case series and a meta-analysis. Utrasound Obstetric Gynecology 2008;31:289-95. [ Links ]
5. Otero JC. Rabdomioma: Tumor del corazón fetal. Diagnóstico Prenatal. Rev Colombiana Ginecol Obstetr 2005;56(2):176-9. [ Links ]
6. Staley B, Vail E, Thiele E. Tuberous Sclerosis Complex: Diagnostic Challenges, Presenting Symptoms, and Commonly Missed Signs. Pediatrics 2011;127(1):117-125. [ Links ]
7. Kaplan J, Ades A, Rychik J. Effect of Prenatal Diagnosis on Outcome in Patients With Congenital Heart Disease. Neoreviews 2005;6:326-331. [ Links ]
8. Co-Vu J, Ivsic T. Fetal Echocardiography to Diagnose Fetal Heart Disease. Neoreviews 2012;10:590-604. [ Links ]
9. Gold A. Freeman J. Depigmented Nevi: The Earliest Sign of Tuberous Sclerosis. Pediatrics 1965;35:1003-05. [ Links ]
10. Hernández J, Morelli J. Birthmarks of Potential Medical Significance. Neoreviews 2003;4:263-69. [ Links ]
11. Jozwiak S, Kotulska K, Kasprzyk-Obara J, Domanska-Pakiela D, Tomyn-Drabik M, Roberts P. et al. Clinical and Genotype Studies of Cardiac Tumors in 154 Patients With Tuberous Sclerosis Complex. Pediatrics 2006;118:1146-51. [ Links ]
12. Tiberio D, Franz D, Phillips J. Regression of a Cardiac Rhabdomyoma in a Patient Receiving Everolimus. Pediatrics 2011;127(5):1335-1337. [ Links ]
13. Krueger DA, Northrup H. International Tuberous Sclerosis Complex Consensus Group. Pediatr Neurol 2013;49(4):255-265. [ Links ]

