










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkActa pediátrica de México
On-line version ISSN 2395-8235Print version ISSN 0186-2391
Acta pediatr. Méx vol.35 n.3 México May./Jun. 2014
Caso de sesión anatomo-clínica
Tumor hepático en un neonato
Liver tumor in a neonate
Carlos López-Candiani1, Eduardo López-Corella2
1 Departamento de Neonatología, Instituto Nacional de Pediatría.
2 Departamento de Anatomía Patológica, Instituto Nacional de Pediatría.
Correspondencia
Dr. Carlos López Candiani
Instituto Nacional de Pediatría
Av. Insurgentes Sur 3700-C
Col. Insurgentes Cuicuilco
CP. 04530, México D.F.
Tel. 1084-0900 Ext. 1352
clopezcandiani@hotmail.com
Recibido: marzo, 2014
Aceptado: abril, 2014
Los tumores son causa poco frecuente de atención en el período neonatal; la incidencia publicada es de 1 a 70 casos por cada 100 000 nacidos vivos,1 por lo que no todos los médicos que atienden a recién nacidos verán uno en su vida; sin embargo, cuando se presenta el caso, es necesario hacer un ejercicio clínico para aproximarnos al diagnóstico. Presentamos el siguiente caso de una recién nacida con tumor hepático para conocer su presentación, su evolución y su diagnóstico definitivo.
CASO CLÍNICO
Antecedentes: madre de 21 años y padre de 28 años de edad, sanos. Producto de segundo embarazo, la madre cursó con infección urinaria tratada con antibiótico no especificado; dos ultrasonidos sin detectar anormalidades. Nació a término en parto vaginal, Apgar 7/9, con peso de 2 740 g y talla de 47 cm.
Padecimiento: al nacer se notó aumento de volumen abdominal con red venosa visible y matidez generalizada. Se envió a hospital pediátrico donde realizaron ultrasonido abdominal e informaron hepatomegalia con patrón heterogéneo y nódulos hiperecoicos. Los estudios de laboratorio evidenciaron trombocitopenia y tiempos de coagulación alargados. Se aplicó vitamina K, plasma y concentrado plaquetario y se refirió a la paciente a tercer nivel.
Al ingreso frecuencia cardíaca de 145 latidos por minuto, frecuencia respiratoria 75 latidos por minuto, tensión arterial de 54/31 mmHg, temperatura: 38°C. Abdomen globoso, lustroso, tenso, con red venosa notable, matidez generalizada; masa palpable de flanco derecho hasta fosa ilíaca que rebasaba la línea media hacia la izquierda, no móvil, bordes bien limitados, dolorosa a la palpación. Resto de exploración física y neurológica normal. Los estudios de laboratorio mostraron: hemoglobina 12 g/dL, 2 500 leucocitos/mm3 con 400 neutrófilos totales y 400 linfocitos totales. Urea: 20.1 mg/dL; creatinina: 1.22 mg/dL; nitrógeno ureico en sangre: 9.4 mg/dL; tiempo de protrombina: 24.9" (33%); tiempo de tromboplastina parcial: 50.7". Bilirrubina indirecta: 10.49 y directa: 1.1 mg/dL. Aspartato aminotransferasa 381 UI/L, alaninoaminotransferasa 53 UI/L, fosfatasa alcalina 90 UI/L, lactato-deshidrogenasa 1 614 UI/L, gammaglutamiltranspeptidasa 160 UI/L. Alfafetoproteína: 49 600 µg/L. Triyodotironina total < 40 ng/dL; tiroxina total: 2.2 mg/dL; hormona estimulante de la tiroides: 0.030 mIU/L; triyodotironina libre: < 1 ng/dL; tiroxina libre: < 0.30 mg/dL. El ultrasonido institucional mostró tumor en ambos lóbulos hepáticos, heterogéneo, que desplazaba al riñón izquierdo y sin poder observar el riñón derecho.
El paciente fue evaluado por cirujanos pediatra y oncólogo pediatra quienes sugirieron el diagnóstico de hemangioendotelioma y síndrome de Kasabach-Merritt por tiempos de coagulación prolongados y trombocitopenia; mencionaron que podría tratarse de hemangioendotelioma versus hamartoma hepático y sugirieron el uso de propranolol y esteroide.
La paciente evolucionó con anuria; el nefrólogo confirmó insuficiencia renal en estadio 2 de la clasificación de la red de lesión renal aguda (Acute Kidney Injury Network-AKIN).2 Se consideró que el tumor podría estar comprimiendo las vías urinarias y las arterias renales con disminución del flujo.
El oncólogo coincidió con la probabilidad de hamartoma o hemangioendotelioma y agregó, como tercera posibilidad, un hepatoblastoma por lo que se solicitó biopsia hepática.
La paciente tuvo deterioro respiratorio con acidosis grave (pH 6.9, pCO2 97.9 mmHg, pO2 44 mmHg y HCO3 21 mg/dL); se le intubó y ventiló con lo que mejoró la eliminación de CO2 (56.1 mmHg) y subió el pH (7.13). Se trató con dopamina y dobutamina, furosemida, vitamina K, transfusión de plasma y concentrados plaquetarios. Se encontró una presión intravesical de 17 cm/H20 y, junto con la anuria, se sospechó síndrome compartimental por lo que se decidió laparotomía descompresiva; hubo bradicardia progresiva que no respondió a las maniobras de reanimación.
Comentario clínico
Clínicamente se hizo un abordaje de una neoplasia en hígado, de gran tamaño y que se acompañó de trombocitopenia y alteraciones de la coagulación. El cuadro 1 muestra una clasificación de tumores hepáticos fetales y neonatales.3 Las tres neoplasias de hígado más frecuentes en la etapa neonatal son hemangioma, hamartoma mesenquimatoso y hepatoblastoma.3 La posibilidad del hemangiotelioma hizo sugerir el uso de propranolol y esteroides.3 Muchos de esos tumores tienen regresión espontánea pero algunos sufren degeneración maligna, por lo que algunos grupos prefieren resecarlos. El hepatoblastoma es un tumor maligno que, dependiendo de su etapa y presentación, puede requerir quimioterapia, cirugía y hasta trasplante hepático.4 El tamaño del tumor y el efecto de masa en el abdomen lo llevó rápidamente a complicaciones que terminaron en el fallecimiento.

Comentario anatomopatológico (Dr. López Corella)
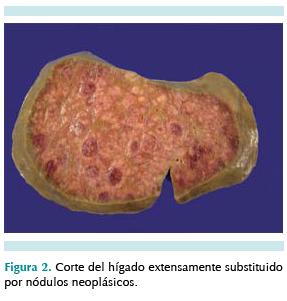
Se recibió cadáver con los diagnósticos de defunción por tumor gigante de hígado, con síndrome compartimental abdominal, insuficiencia cardíaca congestiva e insuficiencia renal aguda. Al momento de la autopsia el abdomen se apreciaba marcadamente voluminoso y distendido y, en la apertura de la cavidad abdominal, resaltaba el gran tamaño del hígado; ocupaba gran parte de la cavidad abdominal y pesaba 510 g cuando el peso esperado en un recién nacido es de aproximadamente 80 g (figura 1). El hígado era firme, con una superficie nodular y en el corte mostraba una gran cantidad de nódulos blanquecinos, rojizos los mayores, que ocupaban la mayor parte del órgano (figura 2). El examen microscópico reveló que el parénquima hepático estaba extensamente infiltrado y substituido por una neoplasia de células pequeñas, de núcleos redondos y uniformes, con muy escaso citoplasma y que alternaban con cordones aislados de hepatocitos y esbozos de conductos biliares residuales (figuras 3 y 4). Aunque no se apreció ninguna indicación de diferenciación neuronal, esta morfología es fuertemente sugestiva de un neuroblastoma y el análisis inmunohistoquímico lo confirmó al resultar positiva para marcadores neuronales (cromogranina, sinaptofisina, enolasa neuronal específica) y negativa para marcadores de tumor neuroectodérmico primitivo periférico/sarcoma de Ewing (CD99) y negativo también para marcadores musculares, linfoides, germinales y epiteliales.



Aunque existen neuroblastomas primarios de hígado, lo más frecuente es que se originen en la médula suprarrenal o en estructuras derivadas de la cresta neural situadas en la línea media como mediastino y ganglios simpáticos abdominales. La autopsia completa mostró, en efecto, que la suprarrenal derecha estaba moderadamente aumentada de tamaño (figura 5) y en el corte se apreció una expansión nodular de esta glándula (figura 6) con la médula ocupada por una neoplasia idéntica a la encontrada en el hígado y que seguramente correspondía al tumor primario. La adrenal izquierda, macroscópicamente normal, también mostró nidos de neuroblastoma en su porción medular que podrían corresponder a un neuroblastoma in situ en este lado, o a una metástasis de la glándula contralateral.


Esta enorme masa en el abdomen seguramente produjo alteraciones hemodinámicas en diversos órganos del paciente que se expresaron en la clínica como insuficiencias cardíaca y renal. Lo más notable en la autopsia fue la evidencia de daño hipóxico/isquémico en el sistema nervioso central: el cerebro mostró zonas de necrosis y hemorragia en la substancia blanca paraventricular, así como leucomalacia periventricular, ambas características de daño hipóxico/isquémico perinatal (figura 7).

Este fue un neuroblastoma congénito, la neoplasia maligna congénita más frecuente seguida, en la mayoría de las series publicadas, por los teratomas y, ya con menos frecuencia, por otros tumores, notablemente leucemias, tumores del sistema nervioso central y sarcomas de tejidos blandos.5 El ultrasonido prenatal pudo haber identificado al tumor hepático, aunque en este caso en particular no era mucho lo que se podía haber hecho en términos de tratamiento. Sin embargo, conviene insistir en la importancia del ultrasonido prenatal porque es capaz de diagnosticar malformaciones, cardíacas y otras, que pueden ser manejadas con un diagnóstico oportuno al nacimiento. Puede asimismo detectar neuroblastomas localizados que sí pueden ser resecados y tienen un buen pronóstico.
El neuroblastoma es una neoplasia errática y contradictoria, que responde bien al tratamiento pero que a la postre exhibe un pronóstico malo en muchos casos. Puede involucionar espontáneamente o madurar a formas benignas; por desgracia situaciones una y otra poco apreciables en la clínica. Forma parte de un espectro de neoplasias relacionadas entre sí, los tumores neuroblásticos periféricos, que va desde el neuroblastoma indiferenciado, como nuestro caso, que no exhibe ningún rasgo morfológico de diferenciación neuronal, pasando por el ganglioneuroblastoma en que una proporción variable de las células hacen núcleos grandes con nucléolos y desarrollan citoplasma de aspecto neuronal hasta el otro extremo; el ganglioneuroma, donde neuronas maduras se encuentran en el seno de un estroma denso característico del producido por las células de Schwann. Entre estos tres escalones de diferenciación hay formas intermedias que han dado lugar a complejos esquemas de clasificación con valor pronóstico. El neuroblastoma y el ganglioneuroblastoma son, en términos generales, tumores malignos mientras que el ganglioneuroma es una condición totalmente benigna. Su presentación clínica suele ser como uno u otro de esos tipos y sólo por excepción se documenta la evolución de una forma a otra más diferenciada; se supone, sin embargo, que esa es la secuencia que siguen todos ellos y que todo ganglioneuroma empezó en algún momento como un neuroblastoma que siguió un derrotero de maduración.
La desconcertante conducta de las formas no maduras, neuroblastoma y ganglioneuroblastoma, ha generado la formulación de esquemas de clasificación y pronóstico basados originalmente en datos histopatológicos (diferenciación y actividad mitótica) y clínicos (etapa clínica y edad del paciente), que se han enriquecido con nuevos marcadores como la amplificación del oncogen MYCN (v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog), la evaluación de la ploidia del ADN y una diversidad de recursos de genética molecular más recientes.6,7
Esto hace posible establecer grupos de histología favorable y de histología desfavorable, con un robusto poder predictivo, que permiten ubicar al paciente dentro de los recursos terapéuticos asequibles (cuadro 2).
En esta clasificación se ve que concurren dos ejes: el morfológico que evalúa el grado de diferenciación y la actividad mitótica y, por otro lado, la edad del paciente. El pronóstico será peor a medida que aumente el grado histológico y la edad del paciente al momento del diagnóstico.7
Pero este tumor plantea una situación particular. Un tumor congénito masivo y no resecable. Se le ha dado el nombre de síndrome de Pepper a un tumor como éste, clásicamente en la adrenal derecha (como nuestro caso) y con metástasis hepáticas masivas que dominan el cuadro clínico.8 Algunos de estos casos encajan dentro de la peculiar forma clínica 4s del neuroblastoma con extensa diseminación multivisceral y a piel y que tienden, con más frecuencia a la habitual, a la regresión espontánea y a un buen pronóstico.
Por otra parte, los neuroblastomas congénitos se cuecen aparte. Son más frecuentemente multicéntricos, de padres más jóvenes y parecen tener un mejor pronóstico cuando son resecables, que no fue el caso de nuestro paciente.9,10 A la luz de esas propuestas podemos considerar que el tumor en la adrenal derecha no es una metástasis sino un ejemplo de un tumor multicéntrico con un neuroblastoma in situ en suprarrenal izquierda y un neuroblastoma en suprarrenal derecha con metástasis masivas en el hígado.
Diagnósticos anatómicos principales.
1. Neuroblastoma congénito en suprarrenal derecha, con metástasis masivas en el hígado (síndrome de Pepper) y con neuroblastoma in situ en suprarrenal izquierda.
2. Daño cerebral hipóxico/isquémico con leucomalacia paraventricular.
Comentario final
La masa de gran tamaño en el hígado, y la falta de evidencia de algún tumor en otra localización, hizo suponer a los clínicos que se trataba de un tumor primario de hígado con las posibilidades diagnósticas mencionadas en el resumen. Sin embargo, nos deja como enseñanza que deben estudiarse todas las posibilidades para llegar a un diagnóstico correcto. En este caso particular no hubiera modificado el desenlace.
REFERENCIAS
1. Davis CF, Carachi R, Young DG. Neonatal tumors: Glasgow 1955-86. Arch Dis Child 1988;63:1075-1078. [ Links ]
2. Mehta R, Kellum J, Shah S, Molitoris B, Ronco C, Warnock D, Levin A, AKINetwork: Acute Kidney Injury Network: Report of an Initiative to Improve Outcomes in Acute Kidney Injury. Critical Care 2007, 11:R31. [ Links ]
3. Isaacs H Jr. Fetal and neonatal hepatic tumors. J Pediatr Surg 2007;42:1797-1803. [ Links ]
4. Thompson PA, Chintagumpala M. Renal and hepatic tumors in the neonatal period. Semin Fetal Neonatal Med 2012;17:216-221. [ Links ]
5. Isaacs H Jr. Fetal and neonatal neuroblastoma: retrospective review of 271 cases. Fetal Pediatr Pathol 2007;26:177-184. [ Links ]
6. Maris JM. Recent advances in neuroblastoma. N Engl J Med 2010;362:2202-2211. [ Links ]
7. Shimada H, Umehara S, Monobe Y, et al. International pathology classification for prognostic evaluation of platients with peripheral neuroblastic tumors. A report from the Children's Cancer Group. Cancer 2001;92:2451-2461. [ Links ]
8. Mabrut JY, Dubois R, Pelizzo G y otros. Abdominal expansion using a polytetrafluoroethylene prosthesis in the treatment of Pepper syndrome. Pediatr Surg Int 2000;16:219-21. [ Links ]
9. Luis AL, Martínez L, Hernández F y otros. Neuroblastomas congénitos. Cir Pediatr 2004;17:89-92. [ Links ]
10. Carlsen NL. Neuroblastomas presenting in the first year of life: epidemiological differences from those presenting at older ages. Cancer Detect Prev 1996;20:251-261. [ Links ]
Nota
Este artículo debe citarse como
López-Candiani C y López-Corella E. Tumor hepático en un neonato. Acta Pediat Mex 2014;35:222-228.

