










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa pediátrica de México
versión On-line ISSN 2395-8235versión impresa ISSN 0186-2391
Acta pediatr. Méx vol.35 no.3 México may./jun. 2014
Caso clínico de interés especial
Síndrome de Johanson-Blizzard. Informe de un caso y revisión de la literatura
Johanson-Blizzard syndrome. Report of a case and review of the literature
Sandra Montserrat Nájera-Villagrana1, Armando Reyes-Cadena2, Angélica León-Hernández3
1 Médico residente del tercer año de Pediatría.
2 Médico adscrito de Pediatría. Instituto Nacional de Pediatría.
3 Médico General. UNAM-FESI, Secretaria de Salud-CNEG y SR.
Correspondencia
Dra. Sandra Montserrat Nájera Villagrana
Instituto Nacional de Pediatría
Av. Insurgentes Sur 3700-C
Colonia Insurgentes Cuicuilco
CP. 04530, México D.F.
monnajera@hotmail.com
Recibido: mayo, 2013
Aceptado: febrero, 2014
RESUMEN
El síndrome de Johanson-Blizzard es una enfermedad con herencia autosómica recesiva; se caracteriza por aplasia o hipoplasia de las alas de la nariz, insuficiencia pancreática exocrina, hipotiroidismo, anomalías dentales y retraso mental. Se han descrito menos de cien casos en el mundo. Presentamos el informe de una niña con todas las características del síndrome con el objeto de facilitar su reconocimiento temprano y de anticipar los graves problemas que conlleva. Actualmente la paciente es vigilada por varios especialistas del Instituto Nacional de Pediatría.
Palabras clave: Johanson-Blizzard síndrome de, hipoplasia nasal, insuficiencia pancreática exocrina, hipotiroidismo, sordera.
ABSTRACT
The Johanson-Blizzard syndrome is an autosomal recessive disease characterized by aplasia or hypoplasia of the wings of the nose, exocrine pancreatic insufficiency, hypothyroidism, dental abnormalities and mental retardation. There are fewer than 100 cases reported in the literature worldwide. We document the case of a girl with all the features of the Johanson-Blizzard syndrome, in order to facilitate its early recognition and anticipate the serious problems associated. Currently the patient is monitored by a multidisciplinary team from Instituto Nacional de Pediatría.
Key words: Johanson-Blizzard syndrome, incidence, nasal hypoplasia, exocrine pancreatic insufficiency, deafness, hypothyrodism.
En 1971 Johanson y Blizzard describieron un síndrome caracterizado por aplasia o hipoplasia congénita de las alas de la nariz, sordera, hipotiroidismo, talla baja, ausencia permanente de los dientes y malabsorción; presentaba un patrón de herencia autosómico recesivo.1,2 Su incidencia se estima en 1 por cada 250 000 recién nacidos. Hasta el año 2013 el número de pacientes descritos en la literatura mundial fue menor de 100 casos.2,3
Las manifestaciones clínicas características de este síndrome son insuficiencia pancreática exocrina, aplasia o hipoplasia de las alas de la nariz, anomalías dentales, defectos del cuero cabelludo, pérdida auditiva neurosensorial, retraso del crecimiento y desarrollo psicomotor, hipotiroidismo, ano imperforado y anomalías genitourinarias.4,5
CASO CLÍNICO
Niña de tres años de edad, con peso de 10.45 kg, en el centil 25, y talla de 85 cm, en centil 5. Tiene retraso en el desarrollo psicomotor con sostenimiento cefálico al año nueve meses de edad, sedestación a los dos años y un mes, gateo a los dos años tres meses; bipedestación a los tres años cinco meses; deambula con ayuda; puede comer por sí sola; requiere ayuda al vestirse; imita a los abuelos y no tiene control de esfínteres. En el área del lenguaje emite balbuceos y se comunica con señas. Exploración física: presenta braquicefalia, frente estrecha con hipertricosis; cejas arqueadas y dispersas; fisuras palpebrales grandes y oblicuas hacia arriba con exposición del aparato lacrimal; puente nasal alto, con nariz corta en pico de loro e hipoplasia de las alas de la nariz (figura 1); filtrum largo y poco marcado; labios delgados; mejillas prominentes; mentón pequeño y puntiforme; orofaringe con paladar alto y estrecho, úvula íntegra y central; microdoncia y diastema (figura 2); pabellones auriculares con implantación adecuada pero acopados; cuello con implantación baja del cabello; tórax normal; abdomen con diástasis de rectos; manchas hiperpigmentadas de bordes irregulares bien definidos lineales que siguen las líneas de Blaschko sobre las caras anterior y posterior del tronco; así como en extremidad superior derecha y cuello anterior, en el hemicuerpo inferior múltiples manchas color café con leche lentiginosas de 3 a 6 mm de diámetro (figura 3); genitales femeninos normales; periné corto; región perianal normal, extremidades íntegras con hipotonía y clinodactilia del quinto dedo.



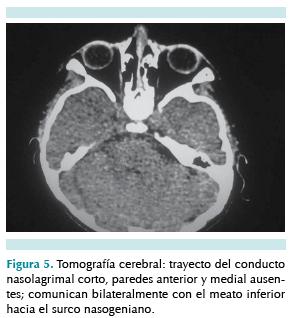
En el ojo izquierdo la niña tiene epífora y secreción purulenta desde los seis meses de edad, por lo que se decidió efectuarle una tomografía axial computada (TAC) de los senos paranasales; el análisis mostró un trayecto del conducto nasolagrimal corto sin paredes anterior y medial; el conducto comunicaba bilateralmente el meato inferior hacia el surco nasogeniano donde se observó hipoplasia de los huesos de la nariz (figuras 4 y 5). Esto confirmó el diagnóstico de dacrioestenosis del ojo izquierdo, motivo por el cual se le practicó dacriointubación y cierre de fístulas laterales.

En un estudio de potenciales evocados auditivos del tallo cerebral se encontraron corticopatía e hipoacusia bilateral; onda V con latencia prolongada y amplitud reducida a 105 decibeles (dB); a 80 dB no se obtuvo ninguna respuesta del lado derecho. Tampoco hubo respuesta a máxima intensidad de iluminación del lado izquierdo.
Se descartó alteración cardiaca por medio de electrocardiograma y ecocardiograma. Al año de edad se le diagnosticó hipotiroidismo y se inició tratamiento con levotiroxina (2.1 µg/kg/día).
Durante su evolución, la paciente presentó síndrome de malabsorción manifestado por diarrea crónica con heces esteatorreicas, detención del crecimiento y distensión abdominal, lo que llevó a realizar pruebas de función pancreática que revelaron una alteración de la función exocrina. Se inició pancreatina a 150 mg VO cada 24 horas. Posteriormente, se diagnosticaron otras alteraciones como acidosis tubular renal y desnutrición crónica agudizada leve, por lo que se requirió un tratamiento con bicarbonato de sodio (8 meq/kg/día) y vitaminas liposolubles.
Para determinar la edad ósea, como seguimiento y control del hipotiroidismo se le tomó una radiografía de la mano, a los tres años 11 meses de edad, que mostró dos núcleos de crecimiento en el carpo y núcleos de crecimiento de las falanges y del radio; ello indicó una maduración ósea que corresponde, aproximadamente, a la de un año para el centil cincuenta; además, las falanges son anchas y los tejidos blandos se encuentran aumentados de volumen, lo que indicó que su maduración ósea estaba atrasada para su edad cronológica (figura 6). Actualmente la paciente se encuentra en seguimiento por diversos especialistas del Instituto Nacional de Pediatría.

DISCUSIÓN
El síndrome de Johanson-Blizzard es una enfermedad hereditaria muy rara del grupo de las displasias ectodérmicas; se caracteriza por múltiples malformaciones y retraso mental.6 Las displasias ectodérmicas son parte del grupo de las genodermatosis congénitas difusas y no progresivas caracterizadas por ausencia o disminución del pelo, dientes, uñas y glándulas sudoríparas y sebáceas, con malformaciones en nariz, pabellones auriculares y labios, así como trastornos del sistema nervioso central.6,7 Dependiendo de las combinaciones existen más de 120 síndromes distintos con diferentes tipos de transmisión hereditaria. Muchos de éstos tienen manifestaciones clínicas que se sobreponen y otras que se distinguen por la presencia o ausencia de un defecto específico.7 Fue descrito por primera vez en 1971 por Johanson y Blizzard: tres pacientes femeninos no relacionados, con aplasia o hipoplasia congénita de las alas de la nariz, defectos ectodérmicos en el cuero cabelludo, ausencia permanente de los dientes, sordera, retraso mental, hipotiroidismo y síndrome de malabsorción; en 1972 Park y sus colaboradores completaron la descripción incluyendo alteraciones urogenitales.1 Antes de estos informes se habían descrito casos clínicos por Morris y Fisher en 1967 y Townes en 1969, quienes los consideraron como ejemplos de enfermedad por deficiencia de tripsinógeno,8 así como por Grand y sus colegas, quienes en 1966 describieron pacientes con síndrome no-clásico de Klinefelter 47,XXY con insuficiencia pancreática, hipotiroidismo, sordera, enfermedad pulmonar crónica, talla baja y microcefalia9. Lumb y Beautyman, en 1952, describieron dos hermanos con fenotipo compatible con dicho síndrome.10 Todas estas descripciones constantes en los diferentes casos clínicos, llevaron a la definición del síndrome de Johanson-Blizzard.
Clínicamente estos pacientes se caracteriza por una nariz inusualmente pequeña con forma de pico de loro, aplasia o hipoplasia de las alas de la nariz, dientes de leche pequeños y malformados, dientes permanentes deformes o ausentes, filtrum largo y estrecho, mentón pequeño y puntiforme, defectos en el cuero cabelludo que van desde cabello distribuido en parches hasta áreas de alopecia con aplasia, cutis congénita subyacente, característicamente en línea media y región occipital.11,12 Los pacientes pueden tener peso bajo al nacer, malabsorción intestinal de las grasas por insuficiencia pancreática exocrina y retraso en el crecimiento durante los primeros años de la vida, lo que redunda en estatura corta. Aproximadamente un tercio de los niños padece hipotiroidismo, que igualmente favorece los retrasos del crecimiento y psicomotor.13
El 80% de los pacientes tienen hipoacusia neurosensorial bilateral congénita que se asocia con ausencia de función vestibular en oído interno estructuralmente normal. En algunos casos puede existir alteración acentuada del lenguaje. También se ha observado retraso mental en aproximadamente 60% de los casos y manchas de color café con leche en 5% de los pacientes.14
Otras malformaciones descritas son: ano imperforado, anormalidades genitourinarias y alteraciones cardiacas como defectos del tabique atrial o ventricular y dextrocardia con transposición de los grandes vasos. Existen casos con deficiencia de la hormona del crecimiento, hipopituitarismo y secreción alterada de glucógeno.14,15
La detención de crecimiento en estos pacientes comienza en el periodo intrauterino y continúa durante la niñez. Se ha sugerido como causa a la hipoplasia del páncreas con insuficiencia exocrina y malabsorción15 debido a que existe un defecto en el tejido acinar del páncreas, con los islotes de Langerhans y conductos preservados, esto provoca reducción casi completa de cimógeno del jugo duodenal con secreción de bicarbonato conservada. Clínicamente el síndrome de malabsorción se caracteriza por diarrea crónica e incapacidad para absorber las grasas. También se ha observado diabetes mellitus en adolescentes, lo que sugiere la naturaleza progresiva de la enfermedad pancreática.16
Estudios genéticos realizados en el 2005 encontraron mutaciones en el gen UBR1 localizado en la región q15q21 del cromosoma 15, que codifican para una ligasa de ubicuitina, fenómeno que se ha relacionado con la alteración de las células acinares del páncreas y de otros órganos.17,18
En 2002 Vieira y sus colaboradores revisaron los 38 casos publicados en el mundo de pacientes con síndrome de Johanson-Blizzard y en los que se mencionaban las características clínicas encontradas en cada uno de ellos. En la mayoría de los pacientes se trató de: síndrome de malabsorción, hipoplasia de las alas de la nariz, lesiones en el cuero cabelludo, sordera, retraso del crecimiento, anormalidades dentales, genitourinarias y anorrectales, hipotiroidismo y anormalidades del conducto lagrimal; las manchas de color café con leche no se observaron con tanta frecuencia.19 En nuestra paciente se observan todas estas anormalidades con excepción de las anormalidades genitourinarias, anorrectales y cardiacas.
El diagnóstico de estos pacientes se puede hacer desde la etapa intrauterina. Se han descrito ultrasonidos obstétricos que muestran dilatación del sigmoides y aplasia de las alas de la nariz, lo cual puede representar el diagnóstico más temprano de este síndrome.20 El primer diagnóstico prenatal de este síndrome por medio de ultrasonido obstétrico fue descrito por Auslander y sus colaboradores en 1999.21
Dado el mecanismo de transmisión es importante referir a los padres para consejo genético, ya que el riesgo de recurrencia después del nacimiento de un individuo afectado es de 25% en cada embarazo.22 Una vez establecido el diagnóstico estos pacientes deben ser evaluados por diferentes especialistas para evitar complicaciones mediante un seguimiento adecuado.22,23
La importancia de informar este caso clínico fue resaltar las diferentes alteraciones congénitas que caracterizan este síndrome, así como la importancia de que el médico general y el pediatra estén familiarizados con este síndrome. Los pacientes con este síndrome requieren diversos estudios en forma oportuna para ofrecer un óptimo tratamiento frente el diagnóstico de ciertas manifestaciones como: síndrome de malabsorción, hipoacusia e hipotiroidismo; esto es de suma importancia ya que permite optimizar el desarrollo psicomotor de estos pacientes.
REFERENCIAS
1. Johanson A, Blizzard R. Syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsortion. J Pediatr 1971;79:982-87. [ Links ]
2. Rezaei N, Sabbaghian M, Liu Z, Zenker M. Eponym: Johanson-Blizzard Syndrome. Eur J Pediatr 2011;170:179-83. [ Links ]
3. Mayerle J, Reis A, Lerch M. Genetic Basis and Pancreatic Biology of Johanson Blizzard Syndrome. Endocrinolo Metab Clin N Am 2006;35:243-53. [ Links ]
4. Ramos S, Ramos H, Ramos R, Peixoto C, Ramos B. Johanson-Blizzard Syndrome. Case Report Braz J Otorhinolaryngol 2010;76(6):794. [ Links ]
5. Amorim K, Batista D, Muniz P, Frassinetti P, Pina G. Johanson-Blizzard Syndrome. A case study of oral and systemic manifestations. Journal Pediatric Otorhinolaringology 2010;5:180-82. [ Links ]
6. Al-Dosari MS, Al-Muhsen S, Al-Jazaeri A, Mayerle J, Zenker M, Alkuraya FS. Johanson-Blizzard syndrome: report of a novel mutation and severe liver involvement. Am J Med Genet A 2008;146 A(14):1875-79. [ Links ]
7. Hurst J, Baraitser M. Johanson-Blizzard Syndrome. Journal of Medical Genetics 1989;26:45-48. [ Links ]
8. Townes PL. Trypsinogen deficiency and other proteolytic deficiency disease. Birth defects. OAS 1972;7(2):95-101. [ Links ]
9. Grand RJ, Rosen SW, Di Sant'Angnese PA, Kirkham WR. Unusual case of XXY Klinefelter's syndrome with pancreatic insufficiency, hypothyroidism, deafness, chronic lung disease, dwarfism and microcephaly. Am J Med 1966;41:478-85. [ Links ]
10. Lumb G, Beautyman W. Hypoplasia of the exocrine tissue of pancreas. J Pathol Bacteriol 1952;64:79. [ Links ]
11. Cheung J, Thomson H, Buncic J, Héon E, Levin AV. Ocular manifestations of the Johanson-Blizzard syndrome. J AAPOS 2009;13(5):512-14. [ Links ]
12. Horbelt C, Aubertin M. A discussion of the physical and oral characteristics of Coffin-Lowry syndrome, Stickler syndrome, and Johanson-Blizzard syndrome. Gen Dent 2009;57(5):468-71. [ Links ]
13. Gershoni B, Lerner A, Braun J, Katzir Y, Iancu TC, Benderly A. Johanson-Blizzard syndrome: clinical spectrum and further delineation of the syndrome. Am J Med Genet 1990;35(4):546-51. [ Links ]
14. Prater J, D'Addio K. Johanson-Blizzard syndrome a case study, behavioral manifestations, and successful treatment strategies. Biol Psychiatry 2002;51(6):515-17. [ Links ]
15. Hoffman W, Lee J, Kovacs K, Chen H, Yaghmai F. Johanson-Blizzard syndrome: autopsy findings with special emphasis on hypopituitarism and review of the literature. Pediatr Dev Pathol 2007;10(1):55-60. [ Links ]
16. Shusshein A, Choi SJ, Silverberg M. Exocrine pancreatic insufficiency with congenital anomalies. J Pediatr 1976;89:782-84. [ Links ]
17. Fallahi G, Sabbaghian M, Khalili M, Parvaneh N, Zenker M, Rezaei N. Novel UBR1 gene mutation in a patient with typical phenotype of Johanson-Blizzard syndrome. Eur J Pediatr 2011;170(2):233-35. [ Links ]
18. Elting M, Kariminejad A, De Sonnaville L, Ottenkamp J, Bauhuber S, Bozorgmehr B, et al. Johanson-Blizzard syndrome caused by identical UBR1 mutations in two unrelated girls, one with a cardiomyopathy. Am J Med Genet A 2008;146(23):3058-61. [ Links ]
19. Vieira M, Lopes V, Teruya H, Guimaraes L, Oliveira L, Duarte C . Johanson-Blizzard Syndrome: The Importance of differential diagnosis in pediatrics. J Pediatr 2002;78(5):433-36. [ Links ]
20. Alpay F, Gül D, Lenk MK, Oğur G. Severe intrauterine growth retardation, aged facial appearance, and congenital heart disease in a newborn with Johanson-Blizzard syndrome. Pediatr Cardiol 2000;21(4):389-90. [ Links ]
21. Auslander R, Nevo O, Diukman R, Morrad E, Bardicef M, Abramov. Johanson-Blizzard syndrome: a prenatal ultrasonographic diagnosis. Ultrasound Obstet Gynecol 1999;13:450-52. [ Links ]
22. Alkohuri N, Kaplan B, Kay M, Shealy A, Crowe C, Bauhuber S, et al. Johanson-Blizzard Syndrome with mild phenotypic features confirmed by UBR1 Gene Testing. J Gastroenterol 2008;14(44):6863-66. [ Links ]
23. Vanlieferinghen H, Borderon C, Francannet H, Gembara P, Dechelotte P. Johanson-Blizzard syndrome. A new case with autopsy findings. Genet Couns 2001;12(3):245-50. [ Links ]
Nota
Este artículo debe citarse como
Nájera-Villagrana SM, Reyes-Cadena A, León-Hernández A. Síndrome de Johanson-Blizzard. Informe de un caso y revisión de la literatura. Acta Pediat Mex 2014;35:212-217.

