










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa pediátrica de México
versión On-line ISSN 2395-8235versión impresa ISSN 0186-2391
Acta pediatr. Méx vol.35 no.2 México mar./abr. 2014
Caso de sesión anatomo-clínica
Lactante con infección pulmonar grave de evolución fatal que se diagnosticó con inmunodeficiencia primaria
Severe fulminant pulmonary infection in an infant diagnosed with primary immunodeficiency
Francisco Javier Espinosa-Rosales1, Cecilia Ridaura-Sanz2
1 Jefe de la Unidad de Inmunología Clínica y Alergia.
2 Jefe del Servicio de Autopsia y Postmortem. Instituto Nacional de Pediatría.
Correspondencia
Dr. Francisco Espinosa Rosales
Jefe de la Unidad de Inmunología Clínica y Alergia
Instituto Nacional de Pediatría
Insurgentes Sur 3700- C.
CP 04530 Ciudad de México
espinosa_francisco@yahoo.com.mx
Recibido: febrero, 2014
Aceptado: marzo, 2014
RESUMEN CLÍNICO
Lactante masculino de 4 meses de edad, primogénito e hijo único de padres jóvenes, aparentemente sanos. El padre refirió tabaquismo, alcoholismo y tener tatuajes. Antecedentes positivos para hipertensión arterial sistémica en tres tíos maternos y diabetes mellitus tipo 2 en abuelos y bisabuelos por ambas ramas. Se negó endogamia, consanguinidad o muertes prematuras en la familia. Paciente originario y residente del Distrito Federal, en casa semiurbanizada, con drenaje y luz, sin agua potable, convivencia con 2 perros y 10 gatos. Miembro de una familia nuclear pequeña, padres con escolaridad básica incompleta y nivel socioeconómico de subsistencia.
Obtenido a término eutócico, con llanto y respiración espontáneos, peso y talla adecuados para edad gestacional (3,150 g y 48 cm para 41 semanas de gestación), Apgar 9/9, egresó como binomio sano a las 24 horas. Onfalorrexis a la semana de vida.
Recibió alimentación mixta desde el nacimiento con fórmula de inicio y leche materna. Inició ablactación a los 4 meses de edad con papillas de frutas. Hitos del desarrollo obtenidos oportunamente hasta su ingreso.
Su esquema de inmunizaciones incluía la aplicación de BCG (dosis única), hepatitis B (2 dosis), pentavalente acelular (1 dosis), rotavirus (1 dosis) y neumococo (1 dosis).
A los 3.5 meses de edad inició con rinorrea hialina y tos en accesos cortos, motivo por el cual acudió a evaluación médica e inició tratamiento ambulatorio con penicilina y paracetamol sin mejoría. Seis días antes de su ingreso se agregó tos en accesos, no productiva, de tono traqueal, disneizante y emetizante, acompañada de rinorrea verdosa, fue llevado ante médico en dos ocasiones más, con tratamientos no especificados, sin observarse recuperación.
El día de su ingreso se agregaron astenia, adinamia, somnolencia, hiporexia, dificultad respiratoria y cianosis, lo que motivó a los padres a buscar atención urgente.
A su ingreso se registraron: peso (5.8 kg, centil 10), talla (62 cm centil 50), perímetro cefálico (41 cm centil 25). Frecuencia cardíaca: 140 latidos por minuto, frecuencia respiratoria de 45 a 80 respiraciones por minuto, presión arterial de 90/56 mmHg, temperatura: 37°C, oximetría de pulso en 85%. Irritable, taquipnéico, con cianosis peribucal y tiraje intercostal. Faringe hiperémica. Campos pulmonares con adecuado murmullo vesicular, sin fenómenos acompañantes. Precordio normal. La exploración de abdomen, genitales y extremidades no fue relevante.
Con la historia de síntomas respiratorios de inicio insidioso y carácter progresivo, que condujeron a dificultad e insuficiencia respiratoria, se decidió su hospitalización con sospecha de síndrome coqueluchoide.
Radiografía de tórax con infiltrado radiopaco homogéneo apical derecho, atelectasia parahiliar izquierda e infiltrados reticulares bilaterales de predominio parahiliar (figura 1).

Por los hallazgos iniciales se comenzó tratamiento con oxígeno en caso cefálico a flujo de 10 L/min, cisaprida, ranitidina, penicilina G sódica cristalina, micronebulizaciones con salbutamol, fluticasona, esteroide sistémico con metilprednisolona con sospecha inicial de neumonía asociada con aspiración. Los primeros 3 días durante su estancia en urgencias persistió con taquipnea, dificultad respiratoria, tos traqueal espasmódica en accesos disneizantes, cianozantes y emetizantes, así como dependencia de oxígeno. Se utilizó alimentación enteral por sonda transpilórica y micronebulizaciones frecuentes con broncodilatador. En el control radiográfico se identificó progresión de los infiltrados con imágenes sugestivas de neumonía de focos múltiples, así como patrón intersticial. Se modificó esquema antimicrobiano a ceftriaxona, dicloxacilina, claritromicina y oseltamivir.
Prueba rápida para influenza: negativa.
Prueba para Bordetella pertussis: negativa.
A los 8 días aparecieron lesiones en la cavidad oral sugestivas de candidiasis oral; se agregó al tratamiento nistatina oral.
A los 17 días de internamiento una serie esófago-gastro-duodenal con mecánica de la deglución reportó succión muy débil y fatiga temprana, además de reflujo gastroesofágico espontáneo y constante hasta el tercio superior del esófago. Por sospecha de alergia a la proteína de la leche de vaca se indicaron un hidrolizado extenso de caseína y un procinético.
El día 18 de su internamiento persistían los síntomas respiratorios y empeoraron los infiltrados pulmonares en las radiografías; se suspendió la dicloxacilina y se sustituyó con vancomicina. Debido a la progresión de la insuficiencia respiratoria se intubó el paciente e ingresó a terapia intensiva.
El día 21, por falta de respuesta al tratamiento y al aislamiento de Candida en hemocultivo se cambió el esquema antimicrobiano a anfotericina B, meropenem, vancomicina y oseltamivir.
El día 23, y ante la sospecha de inmunodeficiencia, se solicitaron ELISA para VIH, subpoblaciones de linfocitos, concentración de inmunoglobulinas y complemento y se inició cobertura contra Pneumocystis jirovecci.
El día 25 de internamiento se reportaron inmunoglobulinas IgG 37.5 mg/dL (nl 196-558), IgM 19.4 mg/dL (nl 27-101), IgA < 24.8 mg/dL (nl 4.4-73), IgE < 1.0 UI/mL (nl 0.18-3.76) y se inició tratamiento sustitutivo e inmunomodulador ante la sepsis grave con inmunoglobulina G a dosis de 1 g/kg.
Debido a que en el día 26 de internamiento se reportó una reacción en cadena de la polimerasa (PCR) positiva para influenza H1N1 se inició un segundo curso con oseltamivir.
El día 28 de estancia hospitalaria se reportó citometría de flujo con CD3 1% (2 LT/mL), CD4 < 1%, CD8 < 1%, CD16/56 no se encontró y CD19 96% (321 LB/mL).
Radiografía de tórax sin timo visible e infiltrado apical derecho homogéneo que sugería consolidación y otro infiltrado parahiliar bilateral con patrón reticular (figura 2).

Durante los días 29 a 30 de hospitalización se deterioró la función respiratoria a pesar del soporte de terapia intensiva y falleció (cuadro 1).
Pruebas de funcionamiento hepático, eletrólitos séricos y químicas sanguíneas realizadas durante la hospitalización sin alteraciones.
Cultivo aspirado de cánula tomado el 19/01/10, reportado el 22/01/10 con 100,000 UFC/mL, Stenotrophomonas maltophilia, Enterococcus faecalis.
ELISA para VIH (1 y 2) reportados como negativos.
COMENTARIO CLÍNICO
De acuerdo con los datos del resumen clínico, este paciente no tenía ninguna característica que permitiera clasificarlo como un paciente de alto riesgo al nacer. En la familia no había consanguinidad ni endogamia, antecedentes de muertes prematuras ni otras enfermedades de carácter congénito o con un patrón de herencia identificable aparente.
Antes del padecimiento fue un lactante con adecuada ganancia pondero-estatural y desarrollo psicomotor normal. Aunque su entorno social pareciera conferirle algún grado de riesgo el paciente tenía su esquema de inmunizaciones al corriente, además de que en diversas ocasiones los padres buscaron atención médica por el padecimiento actual, lo que indirectamente habla del involucramiento de los padres en la atención del menor.
Entre los 3 y 4 meses desarrolló un proceso respiratorio insidioso, progresivo y resistente al tratamiento que, finalmente, lo llevó a la muerte en el lapso de 1 mes.
La evolución cínica y los resultados de laboratorio permiten integrar el diagnóstico de una inmunodeficiencia primaria y descartar otras causas de inmunodeficiencia secundaria a esta edad como la infección por VIH. La radiografía sin timo visible, las concentraciones extremadamente bajas de todas las inmunoglobulinas y el resultado de las subpoblaciones de linfocitos con ausencia de linfocitos T y células asesinas naturales (NK), más las cifras normales de linfocitos B, permiten establecer el diagnóstico de inmunodeficiencia combinada grave.
El hecho de que el paciente fuera un varón sugiere que el gen defectuoso es el que codifica para la cadena gamma común del receptor de múltiples interleucinas (IL-2, IL-4, IL-7, IL-9, IL-15 e IL-21) y que es de herencia recesiva ligada al cromosoma X. Es la forma de inmunodeficiencia combinada grave más frecuente y representa más de 50% de los casos reportados mundialmente.1 Dada la ausencia del antecedente en varones de la familia materna no pueden descartarse otros defectos genéticos más raros que pueden, sin embargo, provocar este patrón en las subpoblaciones de linfocitos: defectos en los genes JAK3, CD45, y coronina-1A, todos ellos de herencia autosómica recesiva.1,2,4
El patrón observado en la citometría de flujo permite descartar razonablemente otras etiologías de inmunodeficiencia combinada grave como son la deficiencia de adenosina-desaminasa (ADA) o purina-nucleósido-fosforilasa (PNP), donde por lo general se observa una linfopenia profunda de LT, LB y NK; defecto de IL-7Ra, en la que esperaríamos tener números normales de células NK, los defectos responsables del síndrome de Omenn (RAG1/2, Artemis, Cernunnos, etc.), que cursan con un cuadro muy característico con eritrodermia, eosinofilia y hepatoesplenomegalia, así como la disgenesia reticular donde existe afección de todas las líneas celulares hematopoyéticas, así como otros defectos más raros.1,2,3
Las inmunodeficiencias combinadas graves (Severe Combined Immunodeficiencies, SCDI) se consideran urgencias en pediatría.
Las SCID ocurren por defectos monogénicos que afectan el desarrollo de los linfocitos T, lo que provoca su disminución o ausencia. El hallazgo característico de estas enfermedades es la linfopenia; sin embargo en este caso, en el que sospechamos que el defecto genético fue causado por un defecto (cadena gamma común) que se caracteriza por no tener linfocitos T ni NK, los linfocitos B existentes podían dar cifras normales de linfocitos en la biometría hemática.1
En un lactante de 4 meses de edad la cuenta normal de linfocitos en una biometría hemática varía de 4,000 a 13,500 células/mm3.5 Algunos autores sugieren que cuentas menores a 2,500 células ameritan evaluación extensiva para documentar o descartar una inmunodeficiencia combinada grave.2,6 Este paciente cursó con cifras de normales bajas (4,200-3,300 células/mm3) a bajas (2,200-660 células/mm3) desde su ingreso.
Pueden observarse, en la biometría hemática, concentraciones "normales" o discretamente disminuidas de linfocitos que corresponden en su mayoría, a linfocitos B. La citometría de flujo con anticuerpos marcados con fluorescencia es indispensable para cuantificar las subpoblaciones de linfocitos T, B y NK.
Ante la sospecha de SCID es indispensable solicitar una citometría de flujo para evaluar la concentración en sangre periférica de linfocitos T (LT) (CD3+), LT cooperadores (CD3+/CD4+), LT citotóxicos (CD3+/CD8+), células asesinas naturales (NK) (CD16+/56+) y linfocitos B (LB) (CD19+).
En este paciente la cuantificación de subpoblaciones de linfocitos fue suficiente para establecer el diagnóstico de SCID. Sin embargo, algunos casos de SCID pueden tener cuentas normales de linfocitos, inclusive mediante citometría de flujo. Y esto generalmente refleja tres escenarios:
1. Linfocitos maternos circulantes en el paciente (que habitualmente son alorreactivos y provocan enfermedad de injerto vs. hospedero [EICH]).1,2,3
2. La mutación en el gen causante de la enfermedad traduce una proteína anormal pero con actividad residual, lo que permite el desarrollo de algunos linfocitos. Sin embargo, estos linfocitos suelen ser pobremente funcionales, oligoclonales y autoreactivos.1,2,3
3. Algunos de los pacientes con SCID pueden requerir de transfusiones de hemoderivados antes de conocerse el diagnóstico. Si los hemoderivados no son irradiados pueden contener linfocitos del donador y generar EICH.1,2,3
Cuando existe la sospecha de SCID, pero no se logra documentar una reducción en el número de LT, es necesario realizar pruebas de activación y proliferación de los linfocitos ante estímulos inespecíficos como la fitohemaglutinina o que activan al LT a través de su receptor (anticuerpos anti-CD3/CD28).
La edad de presentación del caso en discusión coincide con la edad esperada del nadir de concentración de inmunoglobulinas séricas. En lactantes sanos, entre los 3 y 5 meses las inmunoglobulinas maternas transferidas al producto son catabolizadas y descienden hasta niveles de 176 mg/dL.5 Después comienzan a incrementarse paulatinamente hasta lograr las concentraciones normales del adulto alrededor de los 4 a 5 años. A pesar de que esto ocurre en todos los lactantes la mayoría cursa sin problemas este período. Las deficiencias en el número o función de los linfocitos T o B se traducen en deficiencia sostenida en la concentración o en la función de las inmunoglobulinas, así como de las respuestas celulares y provocan un estado de inmunodeficiencia importante a partir de ese momento.2
Las manifestaciones clínicas de este paciente son idénticas a las reportadas frecuentemente en la literatura de pacientes con SCID.1
Los gérmenes identificados en los sitios aislados (Candida spp en cavidad oral y en hemocultivo central, Stenotrophomona y Enterobacter en secreción de cánula, PCR positiva para influenza H1N1) indican susceptibilidad a infecciones por hongos, bacterias y virus, un espectro que comparten algunas inmunodeficiencias, que por la edad de presentación habitualmente son SCID.1-4
Este caso nos muestra que la persistencia, refractariedad y progresión de un problema pulmonar, así como el desarrollo de una infección diseminada por hongos, son datos pivote que nos deben motivar a solicitar una evaluación especializada del sistema inmunitario. Si bien ambos cuadros pueden ser explicados por múltiples factores (i.e. tipo de agente involucrado, virulencia, posible resistencia a antibióticos, así como los efectos derivados del tratamiento antibiótico de amplio espectro y una larga estancia hospitalaria) el retraso del diagnóstico correcto y del tratamiento específico en este caso, como quizá en otros tantos, favoreció un desenlace fatal.
En este paciente se reportó la aplicación de BCG al nacimiento, sin embargo, a diferencia de lo reportado en la mayoría de las series de pacientes con SCID, en países donde se sigue aplicando esta vacuna, no se reportan datos de BCGosis o infección diseminada por BCG.1,7
Al tener la sospecha clínica de SCID debemos iniciar tratamiento intensivo de soporte: mantener en aislamiento protector al paciente (idealmente en un cuarto con presión positiva y con técnicas de esterilidad o cercanas a ellas), profilaxis antimicrobiana, antifúngica y antiviral. Tratamiento enérgico, empírico y de amplio espectro de procesos infecciosos. Cuidados de transmisión de citomegalovirus (CMV), (i.e. serologías para CMV para conocer el estado de la madre o cargas virales de CMV a la madre y al paciente para dar seguimiento); algunos autores sugieren retirar la alimentación al pecho materno ante el riesgo de transmisión de citomegalovirus.2,3
Se debe tener un cuidado especial con el uso de hemoderivados a fin de evitar alosensibilización o infusión de linfocitos viables con el potencial de generar EICH (i.e. solicitar que todos los productos sean irradiados). Se debe administrar inmunoglobulina humana intravenosa a dosis sustitutiva (i.e. 400-600 mg/kg/do cada 21-28 días) o inmunomoduladora (1-2 g/kg/do).2,3
No deben aplicarse vacunas con microorganismos vivos atenuados debido al riesgo de ocasionar infección por el microorganismo utilizado como inmunógeno. Esto es de especial importancia en los primeros meses de vida con la vacunas BCG y contra el rotavirus.2,3
Las únicas posibilidades de ofrecer tratamiento definitivo a estos pacientes son el trasplante de células progenitoras hematopoyéticas (TCPH) o la terapia génica (disponible sólo para algunas formas de SCID en menos de 10 centros en países desarrollados y bajo estrictos protocolos).2,3
Los tres factores más importantes para un pronóstico favorable en un paciente con SCID son:
1. Diagnóstico y tratamiento intensivo de soporte lo más oportuno posible, idealmente antes de la aparición de los signos y síntomas de enfermedad (i.e. complicaciones infecciosas, inflamatorias, autoinmunológicas o neoplásicas).3,6
2. Trasplante de células progenitoras hematopoyéticas antes de los 3.5 meses de vida.3,6
3. Trasplante realizado sin procesos infecciosos.3,6
En el Instituto Nacional de Pediatría se realizó en 2003 el primer trasplante exitoso, de células progenitoras hematopoyéticas, en una paciente con inmunodeficiencia combinada grave. Desde entonces se realizan en promedio 10 trasplantes por año en pacientes con formas graves de inmunodeficiencia primaria.
Desafortunadamente, la tasa de supervivencia en nuestros pacientes es de alrededor de 50% después de un año del trasplante, a diferencia de lo que se reporta en otros países donde las tasas globales de supervivencia rebasan el 70%.8 La problemática especial que viven los pacientes en nuestro país es, en primer término, debida a las complicaciones por la aplicación de vacunas de gérmenes vivos atenuados, específicamente la enfermedad diseminada por BCG. Otros factores que explican la menor tasa de supervivencia en nuestra población son, entre otros, la ausencia de un registro nacional de donadores de células progenitoras hematopoyéticas, lo que dificulta y retrasa en muchos casos por meses a años, la disponibilidad de un donador compatible. Los bancos de sangre de cordón umbilical en nuestro país son pocos y la infraestructura en los existentes es muy limitada, a lo que hay que sumarle que la comunicación entre las diferentes instituciones (por ej. IMSS y SS) es muy deficiente. Además, la mayoría de los pacientes que sufren de estos padecimientos genéticos no tiene hermanos u otros parientes en primer grado genéticamente compatibles.
El desconocimiento del médico de primer contacto de que un niño que sufre infecciones graves o recurrentes puede tener una inmunodeficiencia primaria se traduce, por lo general, en un grave retraso diagnóstico y complicaciones como desnutrición y daño permanente en los órganos que han sufrido infecciones, complicando aún más el tratamiento definitivo y disminuyendo gravemente la posibilidad de supervivencia.
Debido a que los pacientes con SCID no tienen, en la mayoría de las ocasiones, signos clínicos que hagan sospechar sobre el diagnóstico hasta que se presentan infecciones graves, se ha establecido que este grupo de padecimientos cumplen los criterios para ser diagnosticados mediante pruebas de cribado (i.e. tamiz neonatal):
1. Su prevalencia lo justifica; estimada en 1:50,000 recién nacidos vivos.6,9
2. Son difíciles de reconocer mediante aspectos clínicos; habitualmente las manifestaciones son derivadas de la susceptibilidad a procesos infecciosos, autoinmunidad, inflamación o cáncer.6,9
3. Si no se diagnostican y tratan oportunamente pueden producir complicaciones graves o la muerte; las SCID no tratadas son letales en más de 95% de los casos durante los primeros 2 años de vida.6,9
4. El diagnóstico y el tratamiento tempranos modifican el pronóstico; las tasas actuales de supervivencia son mayores de 90% si se tratan antes de los 3.5 meses de vida.6,9
5. Existe una prueba aceptable, sensible, específica, económica y válida disponible; la cuantificación de los círculos de escisión de la recombinación del receptor de células T (TRECs).6,9
Actualmente, en algunos lugares en Estados Unidos efectúan cribado poblacional para SCID en todos los recién nacidos como parte del tamizado neonatal. Esta estrategia ha permitido identificar pacientes con SCID antes de que desarrollen signos y síntomas derivados de las complicaciones antes mencionadas; esto ha permitido ofrecer tratamiento curativo en óptimas condiciones, logrando tasas de supervivencia mayores de 95%.6,9
En México, los primeros esfuerzos por llevar a cabo esta estrategia de cribado poblacional neonatal para SCID son una de las líneas de investigación de la Unidad de Investigación en Inmunodeficiencias del Instituto Nacional de Pediatría en colaboración con el Gobierno del Estado de Tabasco.
CONCLUSIÓN
Se trató de un lactante varón con inmunodeficiencia combinada grave, que desarrolló un cuadro infeccioso por múltiples patógenos que lo llevaron a la muerte antes de poder realizar el tratamiento curativo.
Hallazgos de anatomía patológica
En el estudio post mortem las alteraciones más importantes se encontraron en el sistema inmunológico y en el aparato respiratorio. El timo en el mediastino anterior tenía menos peso pero conservaba su aspecto lobulado (figura 3). Histológicamente, las folias eran delgadas separadas por bandas gruesas de tejido fibroso vascularizado, con pérdida parcial de la demarcación corticomedular por una disminución importante de la población linfoide cortical (figura 4), constituida predominantemente por LT (CD3+) y escasos LB (CD 20+) (figura 5). La porción medular estaba constituida por células fusiformes de aspecto epitelial y no había corpúsculos de Hassall.



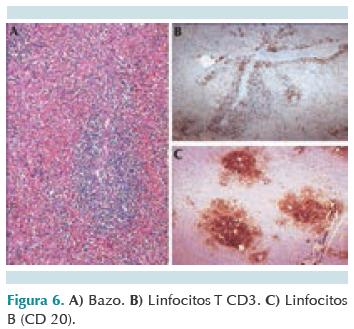
No se encontró tejido linfoide en la fosa amigdalina, placas de Peyer ni en folículos linfoides del apéndice cecal. En el bazo, la pulpa blanca mostró despoblación importante del tejido linfoide. No había formación de folículos linfoides y sólo se conservaban linfocitos dispersos alrededor de las arterias, predominantemente LB (CD20+) y ocasionalmente LT (figura 6). Los ganglios linfáticos del mesenterio eran muy pequeños e histológicamente sólo mostraron grupos escasos de LB agrupados y LT en las porciones paracorticales (figura 7). Estas alteraciones, en general, son las esperadas en un caso de SCID. Sin embargo, la morfología del timo no es la imagen típica de displasia que vemos en los casos de SCID clásicos, ya que se conserva la estructura de las folias y hay linfocitos T en la cortical, lo que sugiere alguna de las formas menos frecuentes de inmunodeficiencia combinada.10

El cuadro respiratorio persistente en este paciente y rebelde a los tratamientos instituidos se explica por los hallazgos de árbol respiratorio. Los pulmones se encontraron aumentados de peso, consolidados, no ventilados, en forma difusa en todos los lóbulos. Histológicamente, había un daño extenso con bronquiolitis y neumonía organizada (figura 8). Los alvéolos estaban ocupados por material proteináceo y detritus celulares, las paredes alveolares estaban revestidas de neumocitos en regeneración y múltiples células gigantes multinucleadas con inclusiones eosinófilas, en el citoplasma, de tamaño y número variables (figura 9).


Esta morfología corresponde a daño citopático ocasionado por paramixovirus (parainfluenza o sincicial respiratorio). En la historia clínica se menciona la infección por virus de la influenza N1H1, lo cual no se puede ni descartar ni confirmar dado que las alteraciones histológicas producidas por este agente son inespecíficas y no permiten su identificación con base en hallazgos morfológicos.11,12
Además de lo anterior se encontraron algunos bronquios con la pared destruida con hifas y esporas de Candida extendiéndose al parénquima vecino (figura 10). Focalmente hay estructuras quísticas PAS + y Groccott + que corresponden a Pneumocystis.11,12 (figura 11).


La combinación de estos tres agentes oportunistas es la esperada en casos de SCID.
Otros hallazgos de la autopsia fueron una esteatosis hepática generalizada ocasionada, seguramente, por malnutrición aguda, atrofia cerebral con daño neuronal hipóxico y necrosis subcortical incipiente de tipo isquémico.
Comentario clínico posterior a los hallazgos de la autopsia
A pesar de que el timo no muestra los hallazgos clásicos descritos en una inmunodeficiencia combinada grave, sí coincide con los hallazgos reportados en el modelo animal responsable de la SCID ligada al cromosoma X: timos con número variable de linfocitos T que incluso pueden llegar a tener corpúsculos de Hassal visibles.13-15 El resto de los hallazgos de la autopsia coinciden con lo descrito en todas las formas de inmunodeficiencia combinada grave.1,14,15
En 1986 Nezelof intentó establecer una correlación entre los hallazgos anatomopatológicos en el timo y la gravedad de las inmunodeficiencias; sin embargo, y debido a la gran variedad en el fenotipo que cada defecto genético muestra, hemos aprendido que no es posible identificar o predecir el defecto genético de los pacientes con base en los hallazgos morfológicos.16
Agradecimiento
Los autores agradecen la ayuda del doctor Armando Partida Gaytán en la redacción y corrección editorial del manuscrito.
REFERENCIAS
1. Puck JM. X-linked Severe Combined Immunodeficiency, Capítulo 9. En: Primary Immunodeficiency Diseases: A molecular and genetic approach. 2nd edition, Oxford University Press, 2007. pp. 123-136. [ Links ]
2. Griffith LM, Cowan MJ, Notarangelo LD, Puck JM, et al. Improving cellular therapy for primary immune deficiency diseases: Recognition, diagnosis and management. J Allergy Clin Immunol 2009;124:1152-60. [ Links ]
3. Bonilla FA1, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Kobrynski LJ, Levinson AI, Mazer B, Nelson RP Jr, Orange JS, Routes JM, Shearer WT, Sorensen RU. American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol 2005;94(5)( Suppl 1):S1-S63. [ Links ]
4. Al-Herz W, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Frontiers in immunology 2011;4:1-26. [ Links ]
5. Tschudy, Megan M, and Kristin M. Arcara. The Harriet Lane Handbook: A Manual for Pediatric House Officers. Philadelphia, PA: Mosby Elsevier, 2012. [ Links ]
6. Verbksy J, Thakar M, and Routes J. The Wisconsin approach to newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 2012;129:622-7. [ Links ]
7. Antaya RJ1, Gardner ES, Bettencourt MS, Daines M, Denise Y, Uthaisangsook S, Buckley RH, Prose NS. Cutaneous complications of BCG vaccination in infants with immune disorders: two cases and a review of the literature. Pediatr Dermatol 2001;18:205-9. [ Links ]
8. Olaya V.A. Transplante de células progenitoras hematopoyéticas en pediatría. Principios básicos. Ciudad de México: Manual Moderno, 2012. [ Links ]
9. IDF SCID Newborn Screening Campaign http://primaryimmune.org/idf-advocacy-center/idf-scid-newborn-screening-campaign/Immune Deficiency Foundation, 40 West Chesapeake Avenue, Suite 308, Towson, Maryland 21204 Email: info@primaryimmune.org Phone: 1.800.296.4433 ©2013 All Rights Reserved. [ Links ]
10. Shearer WT, Fisher A. The last 80 years in primary immunodeficiency. How far have we come, how far need we go? J Allergy Clin Immunol 2006;117:748-52. [ Links ]
11. Rosati LA, Leslie KO. Lung infections. Capítulo 6, páginas: 97-180. Leslie/Wick editors Practical Pulmonary Pathology: A diagnostic approach Churchill Livingstone, 2005. [ Links ]
12. Travis, Colby, Koss. Non-Neoplastic Disorders of the lower respiratory Tract AFIP 2002 Lung infections. Capítulo 12, pp. 539-660. [ Links ]
13. Snyder WP, Kazacos AE and Felsburg PJ. Histologic characterization of the Thymus in Canine X-Linked Severe Combined Immunodeficiency. Clinical Immunology and Immunopathology 1993;67:55-67. [ Links ]
14. Hale LP, et al. Abnormal development of thymic dendritic and epithelial cells in human X-linked severe combined immunodeficiency. Clinical Immunology 2004;110:63-70. [ Links ]
15. Pearse G. Normal structure, function and histology of the thymus. Toxicologic Pathology 2006;34:504-14. [ Links ]
16. Gosseye S, Diebold N, Griscelli C, Nezelof C. Severe combined immunodeficiency disease: a pathological analysis of 26 cases. Clin Immunol Immunopathol 1983;29:58-77. [ Links ]

