










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa pediátrica de México
versión On-line ISSN 2395-8235versión impresa ISSN 0186-2391
Acta pediatr. Méx vol.35 no.1 México ene./feb. 2014
Caso de sesión anatomo-clínica
Enfermedad injerto contra huésped en un paciente con leucemia granulocítica crónica con trasplante de células progenitoras hematopoyéticas
Graft versus host disease in a patient with chronic granulocytic leukemia who received an hematopoietic progenitor cell transplant
Marta Zapata-Tarrés,1 Eduardo López-Corella,2 Martín Pérez-García,3 Roberto Rivera-Luna4
1 Servicio de Oncología.
2 Departamento de Patología.
3 Servicio de Trasplantes.
4 Subdirección de Hemato-Oncología. Instituto Nacional de Pediatría, México DF.
Correspondencia
Dr. Roberto Rivera Luna
Subdirector de Hemato-oncología.
Instituto Nacional de Pediatría
riveraluna@terra.com.mx
Recibido: noviembre, 2013
Aceptado: enero, 2014
RESUMEN DE LA HISTORIA CLÍNICA
Niño de 6 años 9 meses, originario del Estado de México, sin antecedentes de importancia. Padres de 32 años, con educación media superior, comerciantes; tiene un hermano de 10 años y una hermana de 4 años. Cuenta con cartilla de vacunación completa. Dos meses atrás inició con fiebre, epistaxis y esplenomegalia. En el Hospital Materno Infantil de Toluca la biometría hemática reportó: hemoglobina 8.8 g/dL, hematócrito 24%, leucocitos 230,500/mm3, neutrófilos 21%, bandas 34%, linfocitos 9%, monocitos 7%, blastos 14%, basófilos 11%, eosinófilos 4%. Un análisis reveló: creatinina sérica y pruebas de función hepática normales.
En la exploración física se detectó esplenomegalia de 15 cm. Un frotis de sangre periférica mostró datos compatibles con leucemia granulocítica crónica. El aspirado de médula ósea reveló hipercelularidad, 5% de blastos y proliferación de todas las series, BCR/ABL en ABC 27,253 copias. Se trató inicialmente con imatinib (inhibidor de las tirosin cinasas) a dosis de 450 mg/m2. El paciente fue referido al Instituto Nacional de Pediatría para valoración por los médicos del servicio de Trasplante de Células Progenitoras Hematopoyéticos.
A la llegada del paciente se observó: palidez de tegumentos y dermatitis facial secundaria a imatinib. El día de la valoración en el servicio de Trasplante de Células Progenitoras Hematopoyéticos el paciente estuvo recibió 300 mg de imatinib al día; no existen datos de actividad tumoral. Se consideró apto para trasplante de células progenitoras hematopoyéticas en caso de tener donador compatible y se planearon estudios de extensión y HLA.
El resultado de HLA fue compatible al 100% con el hermano sano de 10 años. El resultado por RT-PCR del aspirado de médula ósea t(9,22) fue positivo. El paciente continuaba recibiendo tratamiento con imatinib.
Ingresó al servicio de Trasplante de Células Progenitoras Hematopoyéticos e inició tratamiento de preparación con busulfán. Se colocó un catéter en la arteria subclavia izquierda. Al octavo día se infundió injerto (dosis celular 1.6 x106/kg de peso); sin embargo, debido a un coágulo la cantidad de células no fue suficiente, por lo que se trató nuevamente con una cosecha de donador 100% compatible, sin complicaciones (CD34+ 7.7 x 106/kg de peso). A los diez días del trasplante, el paciente tuvo injerto mieloide, por lo que se suspendió el factor estimulante de colonia de granulocitos. Se decidió su egreso por mejoría.
El día +20 el niño se encontraba asintomático y en buen estado general, alerta y reactivo; se continuó tratamiento con ciclosporina, fluconazol, TMP/SMZ, aciclovir y omeprazol.
Ocho días después de la consulta acudió por haber tenido un pico febril de 38°C. Mencionó que dos días antes tuvo lesiones cutáneas en el cuello y las axilas, sin otros síntomas. A su ingreso tenía frecuencia cardiaca de 120 por minuto; fiebre de 38.2°C; eritema cutáneo generalizado, descamación, sin prurito; costras por úlceras en la región occipital; pulsos distales, llenado capilar de dos segundos. Aparentemente no había evidencia de infección.
El paciente fue valorado por los médicos del servicio de infectología y se decidió prescribirle antibiótico: ceftriaxona y dicloxacilina. Se reinició la ciclosporina a dosis de 6 mg/kg/día, metilprednisolona 2 mg/kg/día y tacrolimus tópico. Se hospitalizó debido a los datos clínicos de enfermedad injerto contra huésped en la piel y para confirmar el diagnóstico.
El servicio de Dermatología diagnosticó una dermatosis diseminada que afectó, principalmente, pliegues del cuello, axilas y región infraclavicular, constituida por descamación, escama gruesa blanquecina, que se desprendía fácilmente, exantema pruriginoso macular de predominio en las piernas, máculas de 5 mm. Este problema evolucionó en tres días; se tomó biopsia de piel. Se diagnosticó enfermedad de injerto contra huésped clínicamente aguda.
Los estudios de virología mostraron: Epstein Barr Virus (EBV), VCA IgM e IgG negativos, EBV EA negativo, EBV EBNA negativo, herpes simple virus IgM E IgG negativos, parvovirus B19 IgM E IgG negativos.
Al día siguiente, luego de ingerir alimentos, refirió dolor epigástrico tipo cólico, sin evacuaciones; un labstix mostró sangre. El ultrsonido abdominal reveló inflamación de íleon, colon y apéndice con menos de 10 mL de líquido libre; asas intestinales con grosor incluso de 6 mm. Se concluyó que el estudio sugería colitis. Se le dejó en ayuno y con analgésico. Se agregó metronidazol y se dio vitamina K debido a que tenía tiempo de protrombina prolongado. Continuó en ayuno por dolor abdominal. tuvo edema facial y en ambas manos. La uresis debajo de valores normales. Se le agregó al tratamiento prednisona y sertralina.
Seis días después continuaba con dolor abdominal, náusea, vómitos e incremento del gasto fecal con evacuaciones de consistencia disminuida. Se detectó hipoalbuminemia de 1.5 y examen general de orina con proteinuria. Un estudio coproparasitoscópico fue positivo para Blastocystis homini y antígeno para Aspergillus negativo. Se inició nutrición parenteral. Se le administró dosis de infliximab dosis-respuesta.
Tres días después tuvo una crisis convulsiva caracterizada por movimientos clónicos, supraversión de la mirada y chupeteo con duración de 30 segundos. Un estudio de electrólitos séricos mostró hiponatremia, hipocalcemia e hipomagnesemia. Posteriormente tuvo nuevas crisis convulsivas de tipo parcial compleja, con deterioro neurológico, Glasgow de 7. Por este motivo se realizó intubación orotraqueal y se le prescribió difenil hidantoína. Se agregaron meropenem y anfotericina B liposomal. La tomografía de cráneo descartó edema y hemorragia cerebral. La resonancia magnética cerebral mostró zonas de edema de tipo vasogénico corticosubcorticales en las regiones parietooccipitales, esto hizo pensar en un síndrome de encefalopatía posterior reversible y atrofia cerebral corticosubcortical. Se suspendieron la difenil hidantoína y la metilprednisolona.
Se aumentó la dosis de ciclosporina y se inició hidrocortisona a dosis de 50 mg. El hemocultivo fue positivo a cocos grampositivos, por eso se agregó vancomicina. Un día previo a su muerte tenía hipoalbuminemia, edema generalizado e hipotérmico. Tuvo hipocalemia de 2.4. El electrocardiograma mostró aplanamiento de ondas T. Debido a que tuvo hiperglucemias hasta de 367 mg se le administró una infusión de insulina. Se suspendió la hidrocortisona y se inició metilprednisolona.
El día de su muerte tuvo desaturación incluso de 60%, se aspiró y se le dio ventilación con presión positiva, con lo que mejoraron la saturación y el patrón ventilatorio. La gasometría reveló pH 7.43, pCO2 30, pO2 61, HCO3 20, CO2T 21. Tuvo hematuria, sangrado por la cánula orotraqueal y una evacuación melénica. Continuó con eventos de desaturación e inestabilidad hemodinámica con bradicardia hasta llegar a la asistolia, sin respuesta a las maniobras de reanimación.
COMENTARIO CLÍNICO
El análisis del caso, para fines académicos, puede separarse en la forma de abordar el diagnóstico de la leucemia granulocítica crónica, la descripción del TCPH y el evento final de enfermedad de injerto contra huésped aguda que representa dificultades para establecer el diagnóstico y dar el tratamiento.
El paciente inició su padecimiento con síndrome febril y hemorragíparo. La asociación de estos dos síndromes obligó a realizar una biometría hemática. A este cuadro se agregó una masa abdominal en el hipocondrio izquierdo.
¿Qué se podía sospechar en este paciente? El pediatra, ante un paciente con una masa abdominal (aunque esté asintomático), debe referirlo a un oncólogo pediatra para determinar la secuencia de estudios lo menos invasivos que permitan integrar el diagnóstico. La biometría hemática mostró anemia de 8.8 mg/dL, leucocitos 230,500, blastos 14%; no se mencionan las plaquetas.
Antes de realizar el aspirado de médula ósea deben descartarse las urgencias oncológicas; en este caso, eran principalmente el síndrome de lisis tumoral, una masa mediastinal y complicaciones por la hiperleucocitosis, como: hipoxemia y crisis convulsivas. Esta última es poco frecuente en los niños con leucemia granulocítica crónica; sin embargo, el diagnóstico más probable en un niño con hiperleucocitosis es una leucemia aguda. La basofilia es un dato frecuente en leucemia granulocítica crónica.
Con estos datos debe realizarse un aspirado de médula ósea. Sin embargo, en la leucemia granulocítica crónica el análisis del frotis de sangre periférica puede ser suficiente para su diagnóstico inicial. En un principio, el tratamiento debe enfocarse a la citorreducción, lo que puede hacerse con hidroxiurea y busulfan. En la actualidad, el imatinib, inhibidor directo de la tirosin cinasa, logra la remisión completa en más de 95% de los pacientes.
Tomando en cuenta los antecedentes de este paciente pueden describirse algunos factores de riesgo para desarrollar leucemia. La edad de los padres se ha relacionado con el cáncer; principalmente leucemia. En el padre, la edad mayor de 40 años se ha vinculado con leucemia, con un riesgo relativo superior a 3. Se han estudiado las condiciones durante el embarazo y la edad materna, sin encontrar alguna asociación específica.
Puesto que el paciente tuvo una leucemia granulocítica crónica, el único factor descrito en la edad pediátrica es la radiación ionizante. No se ha asociado a infección, y es muy raro que aparezca como segunda neoplasia.
La leucemia granulocítica crónica es una enfermedad muy rara en niños: 2 casos por cada 100,000 habitantes, que ocurre en la cuarta y quinta décadas de la vida. En niños se han descrito casos en lactantes, pero 80% se diagnostican después de los 4 años y 60% después de los 6 años.
La leucemia granulocítica crónica se describió por primera vez en 1845 por Bennett. Fue el primer reporte de neoplasia maligna asociada con una anormalidad cromosómica específica. Daley demostró que el fenotipo maligno de la enfermedad es causado por el gen de fusión BCR-ABL, cuyos transcriptos dan lugar a la producción de una tirosin cinasa (PM 210kD) activa, que influye en la adhesión celular a nivel del estroma de médula ósea y que, además, inhibe la apoptosis.
Anteriormente no se había encontrado un tratamiento eficaz contra esta enfermedad, por lo que los pacientes morían alrededor de los tres años de realizado el diagnóstico, producto de una crisis blástica. Fue en la década de 1990, con el desarrollo de inhibidores de tirosin cinasa (mesilato de imatinib), que los pacientes lograron remisiones citogenéticas completas; los seguimientos a siete años mencionan una supervivencia libre de enfermedad de 83% y libre de crisis blástica de 92%.
En la actualidad existe el dilema, incluso en pacientes que tienen donador 100% compatible, si debe o no realizarse un trasplante de células progenitoras hematopoyéticas. A favor, está la supervivencia tan alta libre de evento y, en contra, que el imatinib es un medicamento cuyos estudios clínicos fase III y fase IV aún no tiene resultados en niños.
Desde hace más de 15 años, el trasplante de células progenitoras hematopoyéticas está indicado para esta enfermedad; el acceso al imatinib es más reciente. Surge la pregunta para saber si el pronóstico cambia si se trata antes o después del imatinib, que en realidad retrasa el procedimiento y no sabemos si aumenta o no la morbilidad y la mortalidad. En el Consorcio Europeo de Médula Ósea se señaló que no existen diferencias a 5 años de seguimiento. Así mismo, en la actualidad se desconoce cuál es el tiempo en el que puede suspenderse el medicamento, pregunta que surge sobre todo en niños.
El tratamiento definitivo de este paciente fue con un trasplante de células progenitoras hematopoyéticas alogénico, que se realizó sin complicaciones. Se egresó al día +20. A los 8 días del egreso de la unidad de Trasplante de Células Progenitoras Hematopoyéticos, el paciente regresó con fiebre y lesiones en la piel. Se describen las lesiones, principalmente en pliegues, como descamativas, sin prurito, desprendibles. Las lesiones clásicas que describen incluyen también bulas.
Los diagnósticos diferenciales deben ser reacciones medicamentosas y exantema viral. La biopsia es definitiva en este diagnóstico y, principalmente, se describe infiltración linfocitaria perivascular y, en casos avanzados, separación de la unión dermo-epidérmica. Se agrega dolor abdominal. No se describe la semiología. En el EICH se describen cólicos y también diarrea. El diagnóstico diferencial es una infección intestinal por Clostridium difficile o por citomegalovirus.
El tratamiento profiláctico se basa en ciclosporina, esteroides y tacrolimus. En casos en los que el esteroide se indique a dosis elevadas, como 2 mg/kg, y no exista respuesta, se prescriben anticuerpos monoclonales, como: daclizumab, alemtuzumab o infliximab; como se utilizaron en este paciente, a las tres semanas de su ingreso. El esquema antibiótico indicado estuvo dentro de lo establecido, y el paciente no tenía datos de sepsis en ese momento.
A pesar de esto, el paciente tuvo crisis convulsivas, consideradas inicialmente de origen metabólico. Se descartó hemorragia, situación en la que había que pensar debido a que el paciente tenía 26,000 plaquetas. El niño no tenía fiebre, sin embargo, se considera que hubiera sido importante realizarle una punción lumbar.
Se investigó para citomegalovirus, infección viral que ocurre hasta en 70% de los niños trasplantados; este problema aparece, característicamente, en los primeros 100 días. En este paciente los estudios diagnósticos fueron negativos.
El paciente no respondió al tratamiento. En estos casos debe considerarse qué otros problemas pueden estar ocurriendo, por ejemplo: síndrome urémico-hemolítico o microangiopatía trombótica por ciclosporina. Esta última es una entidad clínica y anatomopatológica caracterizada por: anemia, afectación renal aguda y trombocitopenia causada por una microangiopatía renal predominantemente, pero que puede afectar otros parénquimas, como el sistema nervioso central y el gastrointestinal. En este caso, el paciente tuvo alteraciones compatibles, además de elevación de las concentraciones de urea de 7 hasta 31, aunque la creatinina no se alteró.
La microangiopatía es causada por daño a la microvasculatura debido a inhibidores de la calcineurina, a la quimioterapia o radiación corporal total, o por infecciones. En este momento también puede pensarse en el esquema antibiótico ante la evidencia clínica de deterioro y la falta de evidencia de infección.
Por último, el paciente se deterioró hemodinámicamente quizá debido a una infección no demostrada.
Diagnósticos finales
1. Leucemia granulocítica crónica en remisión.
2. Postrasplante alogénico 100% compatible.
3. Probable microangiopatía trombótica.
4. Enfermedad injerto contra huésped aguda intestinal y piel grado IV.
5. Enfermedad injerto contra huésped cerebral grado IV versus meningitis.
6. Choque séptico-enfermedad injerto contra huésped secundario a una infección no bacteriana.
Causa de muerte: hemorragia pulmonar.
PATOLOGÍA
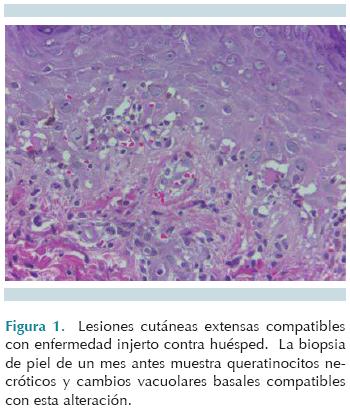
Cuatro horas después de la muerte del paciente se practicó el estudio de autopsia en el Departamento de Patología. Tenía diagnósticos de egreso de hemorragia pulmonar, enfermedad injerto contra huésped en la piel y el intestino y leucemia granulocítica crónica. Mostraba extensas lesiones cutáneas con escaras y descamación; contábamos con la documentación histológica en la biopsia de piel, practicada un mes antes, en la que se apreciaron datos compatibles con enfermedad injerto contra huésped, con queratinocitos necróticos y cambios vacuolares en la porción basal de la epidermis e infiltrados de linfocitos en la unión dermoepidérmica e intraepiteliales. Aunque estos cambios no son específicos ni reacciones a medicamentos, pueden producir alteraciones similares; en el contexto clínico de este paciente puede considerarse el diagnóstico de enfermedad injerto contra huésped (Figura 1. Patología). Esta complicación se explica porque en la autopsia se encontró buena repoblación celular en la médula ósea postrasplante. En ningún órgano se encontró infiltración leucémica.
El cuadro clínico final fue gastroenterológico. En el estómago había algunos cambios focales sugerentes de enfermedad injerto contra huésped, pero el daño más notable estaba en intestino delgado, con una extensísima lesión que afectaba al yeyuno y al íleon pero no al colon. Había necrosis total de la mucosa, con formación de pseudomembranas, fibrina y escasas células inflamatorias mononucleares. (Figura 2. Patología) Puesto que no hubo exudado agudo con neutrófilos, aunque la médula ósea estaba razonablemente poblada, ni movilización de una población de neutrófilos, no se comportaba como una neutropenia. Entonces tenemos una enteritis neutropénica, que en niños suele ser por E. coli o Pseudomonas. En este caso, en el cultivo postmortem se aisló Pseudomonas aeruginosa en el pulmón, hígado, bazo, sangre y placa de Peyer. En el pulmón izquierdo había neumonía hemorrágica multifocal con extensa consolidación. La histología de esta lesión mostraba poca inflamación y extensa necrosis con depósito de un pigmento azulado, característico de infección por Pseudomonas; el antes llamado "bacilo piociánico" (Figura 3. Patología). El paciente tuvo una septicemia por Pseudomonas con el cortejo esperado de complicaciones. No podemos opinar acerca de la participación de la enfermedad injerto contra huésped en la patología intestinal porque, al estar ausente el epitelio de la mucosa, desaparecen los datos diagnósticos de esa alteración.
El sistema nervioso central se estudió con detalle, teniendo en mente las manifestaciones neurológicas en vida. La afectación era poca, con algo de atrofia, daño anóxico muy leve y sin encontrar vasculitis, desmielinizacion o gliosis, por lo que suponemos que la causa de las crisis convulsivas pudo haber sido metabólica o por angioespasmo.
En el timo hubo un hallazgo interesante: era pequeño y sin datos histológicos de atrofia, pero sí de displasia con pérdida linfoide y sin maduración epitelial a los corpúsculos de Hassall. (Figura 4. Patología) Esto se ve en la inmunodeficiencia primaria combinada grave, pero también se ha descrito en la enfermedad injerto contra huésped por transfusión o por trasplante, así como en el síndrome de inmunodeficiencia adquirida. En modelos animales parece ser un fenómeno reversible. Su significado fisiopatológico no es claro.
En resumen, este niño con leucemia granulocítica crónica t(9:22), al que se le hizo trasplante de células progenitoras hematopoyéticas con éxito inició luego con enfermedad injerto contra huésped con lesiones graves en la piel y, quizá, en el tubo digestivo y se complicó con una enteritis neutropénica pseudomembranosa en el contexto de una sepsis por Pseudomonas.
Comentario del servicio de trasplante de células progenitoras hematopoyéticas
En la evolución del trasplante de células progenitoras hematopoyéticas existen tres fases decisivas: la primera es la del pre-trasplante, en la que debe existir una indicación para realizarlo. En este caso el paciente padecía leucemia granulocítica crónica; sin embargo, para que sea apto para el trasplante no es suficiente la indicación de la enfermedad de base, también es necesaria la evaluación orgánica que no contraindique el procedimiento y, por supuesto, una fuente de células progenitoras de sangre periférica, cordón umbilical o médula ósea. Todo ello para ofrecer mayor posibilidad de éxito.
En la segunda fase, la del trasplante, es importante el régimen de condicionamiento. En este paciente fue con busulfán y ciclofosfamida. Existen varios factores a tomar en cuenta para el inicio de la enfermedad injerto contra huésped. En nuestra población, las estadísticamente significativas son: la fuente de sangre periférica, infecciones 3 meses previo al trasplante, infección por citomegaloovirus en el receptor y la dosis celular mayor de de 8.3 x106/kg. Existe más probabilidad de enfermedad injerto contra huésped a mayor dosis celular, por la cantidad de linfocitos que también van infundidos. La otra es la fuente de la que se obtienen las células progenitoras. El mayor riesgo es cuando las células se obtienen de sangre periférica; después, cuando la fuente es médula ósea y menor cuando se utilizan células obtenidas de sangre de cordón umbilical. En este caso no hubo ninguno de estos factores, salvo el hecho de que las células progenitoras hematopoyéticas se obtuvieron de sangre periférica. En este paciente el injerto se efectuó de acuerdo con el tiempo esperado, con evidencia de recuperación en sangre periférica entre 12 y 14 días.
En la tercera fase, la postrasplante, el seguimiento se centra en tres aspectos decisivos: vigilancia de infecciones, sostenimiento del trasplante e inicio de la enfermedad injerto contra huésped, que puede ser aguda o crónica. Este paciente tuvo un síndrome de empalme. Los sitios afectados por la enfermedad injerto contra huésped aguda fueron la piel y el intestino. A mayor inmunosupresión mayor riesgo de complicaciones infecciosas. Entre más inmunosupresores se indiquen mayor será el riesgo, sobre todo de infecciones bacterianas y virales: adenovirus, citomegalovirus, Epstein-Barr, VK o algunas por agentes atípicos. En este paciente, por el tiempo que duró la inmunosupresión, se consideró la posibilidad de un cuadro atípico, como aspergilosis o algún virus que pudo, incluso, causar alteraciones neurológicas. Este caso parece una enfermedad injerto contra huésped en control con un proceso infeccioso mixto bacteriano y viral o bacteriano y micótico secundario a la inmunosupresión.
El hecho de que un paciente se reconstituya hematológicamente, no significa que lo haga inmunológicamente. Esta última puede documentarse luego de 12 o 18 meses, lo que puede explicar las características del timo. Los linfocitos T ya generados por la médula ósea trasplantada migran al timo y hay repoblación; vuelven a formarse los folículos.
En las tres fases del trasplante es indispensable considerar que el paciente y su familia deben estar sujetos a un proceso muy intenso de educación para la salud, porque la detección temprana y la referencia oportunas, lo mismo que los cuidados y el apego al tratamiento inmunosupresor postrasplante, son factores que intervienen en la aparición de complicaciones y su pronóstico final.
COMENTARIO FINAL
En la actualidad, en México, la segunda causa de mortalidad en pacientes entre los 4 y 18 años de edad es el cáncer. Así mismo, nuestro país tiene una de las tasas más altas de cáncer pediátrico en el mundo. El Instituto Nacional de Pediatría posee el servicio de trasplante de células progenitoras hematopoyéticas más grande de la República. En consecuencia, el número de complicaciones que se documentan es elevado. En el caso anterior no sólo se demuestra que no basta que un niño sea trasplantado con éxito en una unidad con experiencia, sino también es necesaria la educación de los padres acerca de los efectos potencialmente mortales que pueden ocurrir. Si a un paciente no se le administra la ciclosporina, se condiciona enfermedad injerto contra huésped y, eventualmente la muerte.
En resumen, se comunica el caso de un niño con leucemia granulocítica crónica que fue tratado inicialmente con imatinib, después con trasplante de células progenitoras hematopoyéticas y que luego inició con enfermedad de injerto contra huésped, lo que lo condujo a la muerte, evento potencialmente evitable con un seguimiento social más estrecho.
REFERENCIAS
1. Castro-Malespina H, Schaison G, Briere J, Passe S, Pasquier A, et al. Philadelphia chromosome-positive chronic myeloid leukemia in children: survival and prognostic features. Cancer 1983;52:721. [ Links ]
2. Gartner JG. Thymic involution with loss of Hassall's corpuscles mimicking thymic dysplasia in a child with transfusion-associated graft-versus-host disease. Pediatr Pathol 1991;11:449-456. [ Links ]
3. Hughes TP, Hochhaus A, Bradford S, Muller MC, Kaeda JS, et al. Long-term prognostic significance of early molecular response to imatinib in neuly diagnosed chronic myeloid leukemia. Blood 2010;116:3758-3765. [ Links ]
4. Lichtman MA. Is There an entity of chemically induced BCR-ABL-positive chronic myelogenous leukemia? The Oncologist 2008;13:645-654. [ Links ]
5. Ortiz-Hidalgo C. Notas sobre la historia de la leucemia. Patología 2013;51:58-89. [ Links ]
6. Rowley JD. A new consistent chromosomal abnormality in chronic muelogenous leukaemia identified by quinacrine fluoresence and Giensa staining. Nature 1973;1:290-293. [ Links ]
7. Vigneri P, Wang JY. Induction of apoptosis in chronic myeloid leukemia cells through nuclear entrapment of BCR-ABL tyrosin kinase. Nat Med 2001;7:228-234. [ Links ]
Nota
Este artículo debe citarse como
Zapata-Tarrés M, López-Corella E, Pérez-Garcí M;a, Rivera-Luna R. Enfermedad injerto contra huésped en un paciente con leucemia granulocítica crónica con trasplante de células progenitoras hematopoyéticas. Acta Pediat Mex 2014;35:45-54.

