










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkCiencias marinas
versão impressa ISSN 0185-3880
Cienc. mar vol.29 no.4 Ensenada Out. 2003
Artículos
Chemical speciation of dissolved lead in polluted environments. A case of study: the Pontevedra Ria (NW Spain)
Especiación química del plomo disuelto en ambientes contaminados. Caso de estudio: la Ría de Pontevedra (NO España)
A. Cobelo-García1*, R. Prego1 and O. Nieto2
1 Grupo de Bioxeoquímica Mariña, Instituto de Investigacións Mariñas (IIM - CSIC), C/ Eduardo Cabello, n°6, 36208 Vigo, España. *E-mail: acobelo@iim.csic.es
2 Departamento de Química Analítica e Alimentaria, Facultade de Ciencias, Universidade de Vigo, Lagoas-Marcosende, s/n, 36200 Vigo, España.
Recibido en octubre de 2001;
aceptado en octubre de 2002.
Abstract
Chemical speciation of dissolved lead was determined at four sampling sites in the Pontevedra Ría (NW Spain) by differential pulse anodic stripping voltammetry (DPASV) with a HMDE. Sampling location was chosen due to its evident anthropogenic influence: one sample was taken in the Lérez River mouth (2.2-salinity) and other three samples were taken in the surroundings of the village of Marín (salinity 30-32). Lead concentrations were 0.64 nM (river sample) and 4.8-21.9 nM (saline samples). Speciation results showed that organic chelates of lead, 88-95% of total dissolved lead, are the dominant species of the metal even at these high concentrations. Two types of lead organic complexing ligands were detected in all samples. The river water sample showed the presence of a strong ligand with a concentration of ~7 nM with a conditional stability constant of K'Pb-L1 ≥ 1011.1, and a weaker ligand (K'Pb-L2 = 108.2) with a concentration of 53.4 nM. The three saline samples showed similar behavior: a strong ligand (K'Pb-L1 ~ 108.6) with concentration ranging from 33.0 to 53.5 nM, and a weaker complexing ligand (K'Pb-L2 ~ 107.5) with concentration ranging from 32.6 to 50.5 nM. All lead-organic ligand complexes (except the strong complex in the river water sample) showed labile behavior in the time scale of the technique.
Key words: Lead, speciation, pollution, rias, Galicia.
Resumen
La especiación química del plomo disuelto se llevó a cabo en cuatro estaciones de muestreo en la Ría Pontevedra (NO de España) utilizando la técnica de voltametría de redisolución anódica de pulso diferencial (DPASV) con un HMDE. La zona de muestreo fue elegida en base a su clara influencia antropogénica: se tomó una muestra en la desembocadura del Río Lérez (salinidad 2.2) y las otras tres se tomaron en las proximidades de la Villa de Marín (salinidad 30-32). Las concentraciones de plomo obtenidas fueron de 0.64 nM (muestra de agua dulce) y 4.8-2.9 nM (muestras salinas). Los resultados de la especiación mostraron que los quelatos orgánicos del plomo, 88-95% del plomo disuelto total, son las especies dominantes del metal incluso a estas elevadas concentraciones. En todas las muestras se detectó la presencia de dos clases de ligandos complejantes del plomo. En la muestra de río se obtuvo un ligando complejante fuerte, de concentración ~7 nM, con una constante condicional K'Pb-L1 ≥ 1011.1, y un ligando más débil (K'Pb-L2 = 108.2) de concentración 53.4 nM. En las tres muestras de aguas salinas se obtuvieron resultados muy similares: un tipo de ligando fuerte (K'Pb-L1 ~ 108.6) con una concentracion comprendida entre 33.0 y 53.5 nM, y un tipo de ligando complejante del plomo más débil (K'Pb-L2 ~ 107.5) con concentraciones comprendidas entre 32.6 y 50.5 nM. Todos los complejos plomo-ligando orgánico (excepto el complejo fuerte de la muestra de río) mostraron un comportamiento lábil en la escala de tiempo de la técnica.
Palabras clave: Plomo, especiación, contaminación, rías, Galicia.
Introduction
Estuaries may contain high levels of both heavy metals and organic matter with respect to the open ocean, mainly due to inputs in their margins derived from anthropogenic activities, transported by freshwater and the atmosphere. The irregular Galician coast is very sensitive to the contaminant effects of heavy metals, due to its 1,720-km long littoral and abundant continental inputs carried by rivers and sewage effluents. However, to date there is no detailed information on the metal concentrations and speciation in the Galician rias; only limited studies have been carried out (Guerrero et al., 1988; Vidal-Collazo, 1991; Antelo, 1992; Vidal-Collazo, 1993; Antelo and Arce, 1996; Cobelo-García et al., 1998a; Cobelo-García et al., 1998b).
The distribution and speciation of metals in seawater is influenced by their total concentration as well as by the organic and inorganic ligands present in the dissolved phase and the co-ordination sites in colloidal and particulate matter (Muller, 1996), and by adsorption sites on the surface of phytoplankton cells (González-Dávila, 1995). Metals in the dissolved phase are present in different physico-chemical forms: (i) free hydrated ion (Mz+), (ii) inorganic complexes (MClz-1, MCO3z-2, etc.) and (iii) organic complexes (MLi). The speciation of metals in the aquatic environment is of great importance and interest since several studies have shown that bioavailability of trace elements (with respect to toxicity and biolimitation) is generally related to the free or inorganic metal fraction, rather than to total concentration (Sunda and Guillard, 1976; Anderson et al., 1978; Brand et al., 1986; Lorenzo et al., 2002).
It has been widely reported that complexation by organic ligands plays a very important role in the speciation of a lot of metals in estuarine, coastal and oceanic waters; much work has been published, especially for Zn and Cu (e.g. van den Berg et al., 1986, 1987; Coale and Bruland, 1988, 1990; Bruland, 1989; Muller and Kester, 1991; van den Berg and Donat, 1992; Donat et al., 1994; Moffett, 1995). The information available for lead in oceanic (Capodaglio et al., 1990, 1991) and coastal and estuarine waters (Muller, 1996; Kozelka et al., 1997; Kozelka and Bruland, 1998; Wells et al., 1998) has also shown that an important fraction of dissolved lead is complexed with organic chelating agents. However, yet there is a lack of information on the lead speciation in areas subjected to metallic contamination. The aim of the present study is to reduce the aforementioned lack of information on lead complexation in the marine environment, especially in those areas with evident anthropogenic inputs and where metal concentrations are high when compared to "typical" or background values.
Methods
Study site
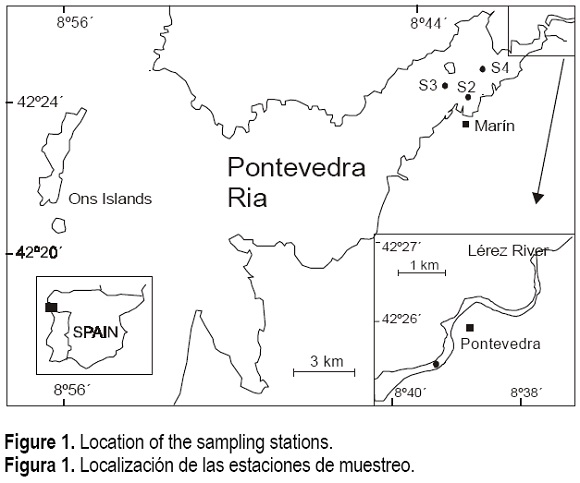
The Pontevedra Ria (fig. 1) is one of the biggest Galician rias and is particularly influenced by anthropogenic activities at: (i) the outflow of the Lérez River due to the proximity of the City of Pontevedra (urban sewage) and a wood and a food processing factory; and (ii) near the Village of Marín, where a wood pulp processing and an electrochemical factory as well as an important shipyard are located. In fact, in this inner part of the ria, in the vicinity of Marín, is where the highest anthropogenic impact onn the ria has been reported (Vilas et al. , 1996). It is therefore likely to find at this points heavy metal contents in the dissolved phase that exceed the average concentrations for coastal and estuarine waters.
Sample collection, filtration and storage
Samples were collected at four sampling sites in the Pontevedra Ria (fig. 1), in March 1999 (samples 1 and 2, S1 and S2) and March 2000 (samples 3 and 4, S3 and S4). Sampling sites were chosen at points likely to present high concentration of metals. Sample 1 was collected at the outflow of the Lérez River and can be considered as river water (2.2-salinity). Samples 2, 3 and 4 were collected in the vicinity of Marín; lead contamination in this ria is located in this area, as was previously reported from the sediment analysis (Herbello et al., 1998).
One liter acid-cleaned polypropylene (PP) bottles were used for sample collection. Immediately after collection, samples were vacuum-filtered through 0.45-µm cellulose acetate filters (Sartorius AG) and stored frozen until analysis. Sample filtration was performed under a laminar flow clean air bench.
Voltammetric equipment
The voltammetric measurement system consisted of a Metrohm 747 VA Stand, a Metrohm 746 VA Trace Analyzer and a Metrohm 695 Autosampler housed inside a clean laboratory. A hanging mercury dropping electrode (HMDE) was used as working electrode; an AgCl/AgCl (KCl sat.) as the reference electrode and a platinum wire as auxiliary electrode. A borosilicate cell was used for total metal concentration; a Teflon cell was used for the titration of samples at their natural pH.
Total dissolved lead determination
Concentration of lead was determined using the method of standard additions, with DPASV detection of the metal deposited at -0.8 V vs. Ag/AgCl (KCl sat.). This determination of total dissolved lead needs the breakdown of organic matter (Scarponi et al., 1996) in order to release the organically complexed metal not detected by DPASV. For this purpose, previous to determination, samples were made 1.4><10-2M in HNO3 and 1.6 > 10-2M in HClO4 reaching a final pH ~1.5 and left standing for several weeks. A similar treatment of samples carried out by Duinker and Kramer (1977) with perchloric acid at lower concentrations (pH 2.7) showed sufficient for a complete release of complexed lead in river and seawater samples.
Total dissolved concentrations of Cd, Zn and Cu were as well determined following the same procedure.
Chemical speciation: ASV-monitored titration with Pb+2
Metals can be found, in the dissolved phase of water, in different physico-chemical forms: as inorganic species -free hydrated ion (Mz+) and complexed with inorganic ligands- or as organic species -forming chelates with the organic matter present-. Lead, as well as other metals, reacts both with inorganic ligands Xi (Cl-, OH-, CO32- etc.) to form MXi complexes, and with generic organic ligands, L. On the other hand, the ligand L takes part in protonation reactions and with the major cations present in the medium (Ca2+, Mg2+, etc.). Moreover, it is assumed that the chelate formed by the lead and the organic ligand has a 1:1 stoichiometry (PbL) and does not participate in side-reactions, therefore the involved equilibrium can be presented as follows (Scarponi et al., 1996):
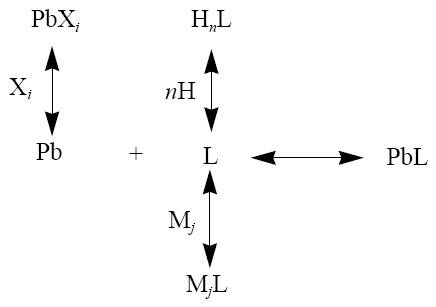
where the stability constant of the complex Pb is given by K=[PbL2-n]/[Pb+2][Ln-]. The constant K is related to the true or thermodynamic stability constant K* by the following relationship: K = K*(ϒPbYL/ϒPbL), where ji is the activity coefficient of species i. If we define [Pb'] as the total lead concentration present in all the inorganic forms (inorganic lead), [L'] as the total organic ligand not complexed with lead, αPb as the inorganic side-reaction coefficient for lead ([Pb+2]=[Pb']/αPb) and c L as the side-reaction coefficient for the inorganic ligand L ([Ln-] = [L']/aL), then we can define a conditional stability constant, K' = [PbL]/[M'][L'] = K/αPbαL, that is related with K by the side-reaction coefficients.
This voltammetric titration methodology is based on the assumption that the ASV technique allows the inorganic concentration of the metal under study ([Pb']) in a sample to be measured. The present speciation methodology (Scarponi et al., 1996; van den Berg, 1982; Ruzic, 1982) is based on the sample titration with the metal under study; the concentration of inorganic lead detected at any point of the titration can be calculated according to [Pb'] = ip/S, where S is the sensitivity of the ASV technique to labile (inorganic) metal and corresponds to the slope of the titration curve after the natural organic ligands have become saturated with metal, and ip is the anodic peak current. The calculations of the speciation parameters, ligand concentrations and conditional stability constants were carried out by transformation of the titration curves using the linearization procedures proposed by Ruzic (1982) and van den Berg (1982). According to this method, the ratio [Pb']/[PbL] is plotted against [Pb']; linearity of this representation over the range of the data was interpreted to indicate complexation of the metal by one class of ligand only. Values for the conditional stability constants with respect to inorganic lead (K') and for the concentration of natural metal binding ligand ([L]) were obtained from the slope and intercept of the [Pb']/[PbL] vs. [Pb'] plot (Ruzic, 1982; van den Berg, 1982; Scarponi et al., 1996):
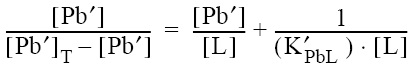
In the presence of two complexing ligands, the Ruzic/van den Berg transformed representation becomes (van den Berg, 1982; Ruzic, 1982; Scarponi et al., 1996):
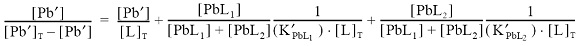
therefore the plot [Pb']/[PbL] vs. [Pb'] is lineal no more, showing a curvature in the representation. For samples 2, 3 and 4 the Ruzic/van den Berg plot showed a curvature of the type indicating metal complexation by two ligands; the speciation parameters (K'PbL1, [L1], K'PbL2, [L2]) were estimated using the approach described by van den Berg (van den Berg, 1984; Scarponi et al., 1996). There was no need to invoke more than two ligands in order to accurately reproduce the titration data.
As was previously mentioned, this methodology of sample titration is based on the ASV technique allowing to measure the inorganic metal concentration. However, there are several situations that may perturb the correct discrimination between the inorganic and organic fractions of metal: (i) the possible reduction of the metal-ligand complex at the applied potential in the deposition step; (ii) the possibility of a kinetic lability of the metal-organic ligand complexes in the diffusion layer (Tuschall and Brezonik, 1981). Both processes would lead to an underestimation of the complexing ligand concentration and the conditional stability constant of the complexes formed.
In order to choose the most suitable deposition potential, minimizing the possible reduction of organically complexed lead, a pseudopolarogram study was carried out for sample 2. A typical sigmoid shape was obtained from -0.60 V to -1.0 V, confirming that the reduction of the metal-ligand complexes did not occur at that range of potentials. From the pseudopo-larogram, a deposition potential of -0.8 V was chosen for all the samples, since it was the minimum overpotential required to reach the reproducible fraction of the pseudopolarogram plateau.
The kinetic contribution to the peak current due to the complex dissociation in the diffusion layer depends on the dissociation rate of the complex (kd) and on the effective time scale of the electrochemical experiment, which is also dependent on the residence time of the complex in the diffusion layer. The thinner is the diffusion layer, the shorter the residence time, the lower the dissociation of the complex and, therefore, the lower the kinetic contribution to the peak current. In order to diminish as much as possible the diffusion layer width, the highest stirring rate (2000 rpm) allowed by the electrochemical device without affecting the stability of the mercury drop during the deposition step was used.
Between 12 and 15 lead additions were spiked to every sample to obtain the voltammetric titration, increasing the lead concentration in the sample from 0 to 175 nM. After each addition, the samples were allowed to equilibrate for 25 minutes, avoiding any kinetic effects in the representations due to non-equilibrium conditions (Ruzic and Nikolic, 1982). The deposition times applied were dependent on the labile Pb concentrations ([Pb']) for every sample and ranged from 2 to 45 minutes. The stripping scan was carried out in the differential pulse mode (DPASV) in the positive direction from -0.80 V to -0.30 V at a rate of 10 mV/s with a pulse width of 50 mV.
Results
The total dissolved lead concentrations found were 0.64 in sample 1 (river water), 8.0 in sample 2, 4.8 in sample 3 and 21.9 in sample 4.
Inorganic lead (Pb') was not detected in sample 1 even at 45-minute deposition times, until several additions were made, as can be seen in figure 2. This is attributed to the presence of a high-stability lead-organic ligand chelate. The concentration of this strong complexing ligand (L1) was estimated (see Bruland et al. , 2000) as the mid-point between the last lead concentration yielding no signal and the lead concentration yielding the first measurable signal, obtaining a concentration ~7 nM (mean of two titrations). In these situations, when no inorganic metal is detected until the ligand is saturated, the product of the conditional stability constant by the ligand concentration is equal or grater than 103 (Scarponi et al., 1996). Ignoring the complexation by weaker organic ligands, a low limit for the conditional stability constant can be estimated as K'1 ≥ 103/(7>10-9) = 1.4X1011. The datapoints obtained for the subsequent Pb additions during the titration showed a curvature indicative of the presence of one or more types of complexing ligands different to the abovementioned. The transformed plot obtained from these data showed a linear behavior and therefore the presence of only one more ligand was assumed. In order to calculate the concentration of this weaker ligand, the concentration of the stronger ligand was subtracted from every titration point, previous to the transformed plot (Bruland et al., 2000). A concentration of 53.4 nM with a log K' = 8.2 was obtained for the weaker ligand. The peak potentials shift towards more anodic values when the total lead concentration increases (fig. 2b), until a constant value is attained corresponding to the peak potential of the free (inorganic) lead, once the second ligand has been completely complexed (saturated). This peak potential shift towards more anodic values indicates that the Pb-L2 complex is labile or quasi-labile in the timescale of the technique used (van Leeuwen, 1987).
In the saline samples, where a higher total dissolved lead was found, an ambient inorganic lead fraction was detected. The concentrations of the inorganic fractions were 0.54 nM (S2), 0.38 nM (S3) and 2.6 nM (S4), obtaining organically complexed lead percentages of 93.3%, 92.1% and 88.2%, respectively. The Ruzic/van den Berg transformed plot for these saline samples showed a typical curvature for the presence of two types of organic ligands, as can be seen in figure 3b for sample 2. As was previously mentioned, the speciation parameters were calculated using the approach proposed by van den Berg (van den Berg, 1984; Scarponi et al., 1996). The concentrations obtained for the "strong" ligands were 53.5 nM (S2), 33.0 nM (S3) and 39.4 nM (S4), whereas the "weak" ligands concentrations were 37.0 nM (S2), 50.5 nM (S3) and 32.6 nM (S4) (table 1). The logarithmic values of the conditional stability constant of the Pb-L1 complex were around 8.6 and, for the Pb-L2, around 7.5. As well as for the river sample (S1), a shift of the peak potentials towards more anodic values was observed (fig. 3c), showing a labile or quasi-labile behavior of these complexes in the timescale of the differential pulse anodic stripping voltammetry using a HMDE.
Discussion
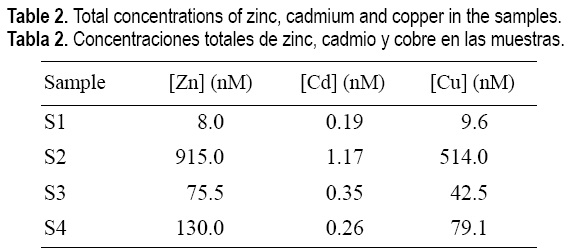
The typical dissolved lead concentrations in coastal and estuarine waters, generally below 1 nM (Cutter, 1991), are widely exceeded in samples 2, 3 and 4, pointing out to the anthropogenic influence in this local area of the inner part of the Pontevedra Ria. These high values are in agreement with the (scarce) existing data that reported lead concentrations in this ria ranging from 2 to 8 nM (Guerrero et al., 1988). This important anthropogenic influence is also evident if we consider the concentrations of Zn, Cd and Cu in those samples (table 2), especially sample S2, where obvious contamination exists. These data are in accordance with the studies carried out in the sediments of the ria, which reported that the highest anthropogenic influence is located in the vicinity of Marín, mainly due to the shipyard and sewage inputs (Vilas et al., 1996; Herbello et al., 1998). However, the metal concentrations of the river water sample were quite lower than the saline samples, showing that the river is not so influenced by contaminating inputs.
Results show that most of lead (> 88% of total dissolved metal) is present as organic complexes in these waters. Two types of organic ligands responsible of that complexation were found in all samples. In the saline samples, the ligands showed a similar behavior, with concentrations of the same order of magnitude and similar conditional stability constants of the Pb-organic ligand complexes - log K'Pb-L1 ~8.6 and log K'Pb-L2 ~7.5, not showing any temporal trend. However, the fluvial sample (S1), in spite of showing also two types of ligands, they showed different characteristics with respect to those of the saline samples. Accordingly, the conditional stability constant of the "strong" ligand was quite greater (log K'Pb-L1 ≥ 11.1) and its concentration lower (~7 nM); the conditional stability constant of the "weak" ligand was greater (log K'Pb-L2 = 8.2), although its concentration was of the same order as the ligands of the saline samples.
It is important to point out that the ligands (except the "strong" one in sample S1) showed a labile or quasi-labile behavior in the timescale of the HMDE electrode, which may lead to an underestimation of the conditional stability constant of the complexes detected, although the ligand concentrations are not affected (Capodaglio et al., 1995). Another consequence of the complex lability is that the labile metal concentration ([Pb']) -and, therefore [Pb+2]- is overestimated, whereas the %Pborganic is underestimated. This labile behavior of the organic complexes when using a HMDE was also reported by Botelho et al. (1994) in a previous study carried out on the lead complexation with organic ligands in contaminated river waters.
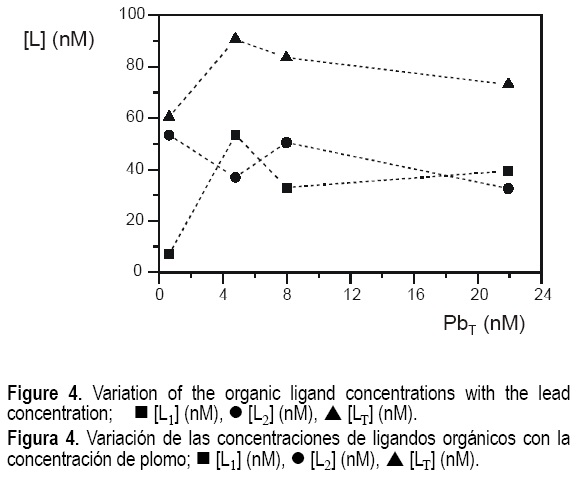
On the other hand, no obvious correlation was found between the total lead concentration and the ligand concentrations, as observed in figure 4. However, a slight decrease in the total ligand concentration when increasing the metal concentration is observed for the saline samples, although the scarce data available do not allow to go deeper in this observation.
Contribution to the knowledge of lead speciation in coastal areas. Comparison with data in literature.
The studies published on chemical lead speciation in estuarine and coastal waters (Muller, 1996; Kozelka et al., 1997; Kozelka and Bruland, 1998; table 3) also reported the presence of two types of organic complexing ligands responsible for, usually, more than 90% of total dissolved lead. This is also confirmed in the present study, suggesting that most of dissolved lead in the estuarine-coastal environment is organically complexed even at high concentrations up to 22 nM. However, there are several differences with the data reported in the literature:
(i) In the present study we have sampled an area where an evident anthropogenic influence exists and therefore the Pb concentrations are one order of maginitude greater than the literature data cited; (ii) ligand concentrations are up to two orders of magnitude greater. One possible reason is the production of complexing exudates from marine phytoplankton exposed to those high metal concentrations (Ahner and Morel, 1995). However, the most probable reason of these high ligand concentrations is that most of the complexing organic matter has (as well as most of the lead present) an anthropogenic origin, from the sewage inputs to the ria. In the sampling area, a wood pulp processing factory and a sewage treatment plant are settled, contributing with high loads of organic matter to this area. In fact, previous studies have shown that sediments in the vicinity of Marín contain the greatest organic load (up to 19%) in the whole ria (Vilas et al., 1996); (iii) the stability constants of the two types of organic complexes found are around 10 times lower; this is coherent if we take into account that at these high lead concentrations the stronger ligands, of lower concentration, are completely saturated before the titration; that is, the analytical windows (van den Berg and Donat, 1992; Bruland et al., 2000) used are different. This makes sense if we compare the strong complexation constants obtained in the Pontevedra Ria (8.5 - 8.6) with the weak constants reported for Narragansett Bay (8.6 - 8.8), the Southern English Coast (8.0 - 9.3) and the San Francisco Bay (8.6), being all very similar.
Acknowledgements
The authors thank R. Beiras and J.I. Lorenzo from the Grupo de Ecoloxía of the Facultade de Ciencias (Universidade de Vigo) for their help with the sampling and determination of pH and salinity. A. Cobelo-García would like to thank the Spanish Ministry of Science and Technology for the financial support (FPI Grant).
References
Ahner, B.A. and Morel, F.M.M. (1995). Phytochelatin production in marine algae. 2. Induction by various metals. Limnol. Oceanogr., 40(4): 658-665. [ Links ]
Anderson, M.A., Morel, F.M.M. and Guillard, R.R.L. (1978). Growth limitation of a coastal diatom by low zinc ion activity. Nature, 276: 70. [ Links ]
Antelo, J.M. (1992). Calidad del agua en las Estaciones de Aforos de los Ríos de Galicia Costa, Años Hidrológicos 89-90 y 90-91. FEUGA, Xunta de Galicia. [ Links ]
Antelo, J.M. and Arce, F. (1996). Características fisicoquímicas das Augas Superficiais. In: Consello da Cultura Galega (ed.), As Augas de Galicia, pp. 351-146. [ Links ]
Botelho, C.M.S., Boaventura, R.A.R. and Goncalves, M.L.S.S. (1994). Interactions of lead (II) with natural river water: part I. Soluble organics. Sci. Tot. Environ., 149: 69-81. [ Links ]
Brand, L.E., Sunda, W.G. and Guillard, R.R.L. (1986). Reduction in marine phytoplankton reproduction rates by copper and cadmiun. J. Exp. Mar. Biol. Ecol., 96: 225-250. [ Links ]
Bruland, K.W. (1989). Complexation of zinc by natural organic ligands in the central North Pacific. Limnol. Oceanogr., 34(2): 269-285. [ Links ]
Bruland, K.W., Rue, E.L., Donat, J.R., Skrabal, S.A. and Moffet, J.W. (2000). Intercomparison of voltammetric techniques to determine the chemical speciation of dissolved copper in a coastal seawater sample. Anal. Chim. Acta, 405: 99-113. [ Links ]
Byrne, R., Kump, L.R. and Cantrell, K.J. (1988). The influence of pH and temperature on trace metal speciation in seawater. Mar. Chem., 25: 163-181. [ Links ]
Capodaglio, G., Coale, K.W. and Bruland, K.W. (1990). Lead speciation in surface waters of the Eastern North Pacific. Mar. Chem., 29: 221-233. [ Links ]
Capodaglio, G., Scarponi, G. and Cescon, P. (1991). Lead speciation in the Antarctic Ocean. Analyt. Proc., 28: 76-77. [ Links ]
Capodaglio, G., Scarponi, G. and Cescon, P. (1995). Speciation of trace metals in seawater by anodic stripping voltammetry: critical analytical steps. Fresenius J. Anal. Chem., 351: 386-392. [ Links ]
Coale, K.H. and Bruland, K.W. (1988). Copper complexation in the northeast Pacific. Limnol. Oceanogr., 33(5): 1084-1101. [ Links ]
Coale, K.H. and Bruland, K.W. (1990). Spatial and temporal variability in copper complexation in the North Pacific. Deep-Sea Res., 37(2): 317-336. [ Links ]
Cobelo-García, A., Prego, R., Fernández-Álvarez, J.M. (1998a). Estudio voltamétrico de los parámetros de complejación del zinc en aguas de las rías gallegas. Series de Química Oceanográfica -Serie II Cuadernos, 2: 17-25. [ Links ]
Cobelo-García, A, Fernández-Álvarez, J.M. and Prego, R. (1998b). In: Universidade de Faro (ed.), Proccedings of the 1st Interdisciplinary Symposium on Estuarine Processes. Faro (Portugal), pp.105-108. [ Links ]
Cutter, G.A. (1991). Trace elements in estuarine and coastal waters: United States studies from 1986-1990. Rev. Geophys., 29: 639-644. [ Links ]
Donat, J.R., Lao, K.A. and Bruland, K.W. (1994). Speciation of dissolved copper and nickel in South San Francisco Bay: a multi-method approach. Anal. Chim. Acta, 284: 547-571. [ Links ]
Duinker, J.C. and Kramer, C.J.M. (1977). An experimental study on the speciation of dissolved zinc, cadmiun, lead and copper in river Rhine and North Sea water, by differential pulsed anodic stripping voltammetry. Mar. Chem., 5: 207-228. [ Links ]
González-Dávila, M. (1995). The role of phytoplancton cells on the control of heavy metal concentration in seawater. Mar. Chem., 48: 215-236. [ Links ]
Guerrero-Pérez, J., Rodríguez-Puente, C. y Jornet-Sancho, A. (1988). Estudio de metales pesados en aguas y sedimentos superficiales en las costas cantábrica y gallega. Informes Técnicos Instituo Español de Oceanografía, n°64. [ Links ]
Herbello, P., Barciela, M.C., Prego, R. and Bermejo, P. (1998). In: Morales and Borrego, (eds.), European Land-Ocean Interaction Studies. Huelva, Spain, p. 62. [ Links ]
Kozelka, P.B., Sanudo-Wilhelmy, S., Flegal, A.R. and Bruland, K.W. (1997). Physico-chemical speciation of lead in South San Francisco Bay. Estuar. Coast. Shelf Sci., 44: 649-658. [ Links ]
Kozelka, P.B. and Bruland, K.W. (1998). Chemical speciation of dissolved Cu, Zn, Cd, Pb in Narragansett Bay, Rhode Island. Mar. Chem., 60: 267-282. [ Links ]
Lorenzo, J.I., Beiras, R. and Nieto, O. (2002). Effects of humic acids on speciation and toxicity of copper to Paracentrotus lividus larvae in seawater. Aquatic Toxicology, 58: 27-41. [ Links ]
Moffett, J.W. (1995). Temporal and spatial variability of copper complexation by strong chelators in the Sargasso Sea. Deep-Sea Res., 42 (8): 1273-1295. [ Links ]
Muller, F.L.L. and Kester, D.R. (1991). Voltammetric determination of the complexation parameters of zinc in marine and estuarine waters. Mar. Chem., 33: 71-90. [ Links ]
Muller, F.L.L. (1996). Interactions of copper, lead and cadmiun with the dissolved, colloidal and particulate components of estuarine and coastal waters. Mar. Chem., 52: 245-268. [ Links ]
Ruzic, I. (1982). Theoretical aspects of the direct titration of natural waters and its information yield for trace metal speciation. Anal. Chim. Acta, 140: 99-113. [ Links ]
Ruzic, I. and Nikolic, S. (1982). The influence of kinetics on the direct titration curves on natural water systems. Theoretical considerations. Anal. Chim. Acta, 140: 331-334. [ Links ]
Scarponi, G., Capodaglio, G., Barbante, C. and Cescon, P. (1996). In: S. Caroli (ed.), Element Speciation in Bioinorganic Chemistry. John Wiley and Sons, Inc., pp. 363-118. [ Links ]
Sunda, W.G. and Guillard, R.R.L. (1976). The relationship between cupric ion activity and the toxicity of copper to phytoplankton. J. Mar. Res., 34: 511-529. [ Links ]
Turner, D.R., Whitfield, M. and Dickson, A.G. (1981). The equilibrium speciation of dissolved components in freshwater and seawater at 25°C and 1 atm pressure. Geochim. Cosmochim. Acta, 45: 855-882. [ Links ]
Tuschall, J.R. and Brezonik, P.L. (1981). Evaluation of copper anodic stripping voltammetry complexometric titration for complexing capacities and conditional stability constants. Anal. Chem., 53: 1986-1989. [ Links ]
van den Berg, C.M.G. (1982). Determination of copper complexation with natural organic ligands in seawater by equilibration with MnO2: I. Theory. Mar. Chem., 11: 307-322. [ Links ]
van den Berg, C.M.G. (1984). Determination of the complexing capacity and conditional stability constants of complexes of copper (II) with natural organic ligands in seawater by cathodic stripping voltammetry of copper-cathecol complex ions. Mar. Chem., 15: 1-18. [ Links ]
van den Berg., C.M.G., Buckley, P.H.M., Huang, Z.Q. and Nimmo, M. (1986). An electrochemical study of the speciation of copper, zinc and iron in two estuaries in England. Estuar. Coast. Shelf Sci., 22: 479-486. [ Links ]
van den Berg., C.M.G., Merks, A.G.A. and Duursma, E.K. (1987). Organic complexation and its control of the dissolved concentrations of copper and zinc in the Scheldt estuary. Estuar. Coast. Shelf Sci., 24: 785-797. [ Links ]
van den Berg, C.M.G. and Donat, J.R. (1992). Determination and data evaluation of copper complexation by organic ligands in sea waater using cothodic stripping voltammetry at varying detection windows. Analyt. Chim. Acta, 257: 281-291. [ Links ]
van Leeuwen, H.P. (1987). Voltammetric titrations involving metal complexes: effect of kinetics and diffusion coefficients. Sci. Tot. Environ., 60: 45-55. [ Links ]
Vidal-Collazo, M.L. (1991). Especiación de Zn, Cd, Pb y Cu en aguas de la ría de Ferrol. Series de Química Oceanográfica - Serie II Cuadernos, 3: 1-54. [ Links ]
Vidal-Collazo, M.L. (1993). Cálculo de las capacidades de complejación de cobre en aguas superficiales de las rías de O Burgo y Ferrol. Series de Química Oceanográfica - Serie II Cuadernos, 2/3: 77-85. [ Links ]
Vilas, F., García-Gil, E., García-Gil, S., Nombela, M.A., Alejo, I., Rubio, B. y Pazos, O. (1996). Cartografía de sedimentos submarinos - Ría de Pontevedra. Xunta de Galicia, 39 pp. [ Links ]
Wells, M.L., Kozelka, P.B. and Bruland, K.W. (1998). The complexation of 'dissolved' Cu, Zn, Cd and Pb by soluble and colloidal organic matter in Narragansett Bay, RI. Mar. Chem., 62: 203-217. [ Links ]

