










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkSalud mental
Print version ISSN 0185-3325
Salud Ment vol.33 n.1 México Jan./Feb. 2010
Actualización por temas
Alteraciones estructurales encefálicas en el trastorno por déficit de atención e hiperactividad: una actualización. Segunda parte*
Structural brain alterations in attention–deficit/hyperactivity disorder: an update. Part two
Luis Guillermo Almeida Montes,1 * Josefina Ricardo–Garcell2, Hugo Prado Alcántara,1 Reyna Beatriz Martínez García1
1 Centro Estatal de Salud Mental, SESEQ.
2 Instituto de Neurobiología UNAM, Campus Juriquilla, Querétaro.
*Correspondencia:
M. en C. Dr. Luis Guillermo Almeida Montes.
Avenida 5 de Febrero 105–Sur,
colonia Virreyes, 76170.
Querétaro, Querétaro. México.
Tel/Fax: (01–442) 224–2487.
E–mail: almeidal@prodigy.net.mx
DISCUSIÓN
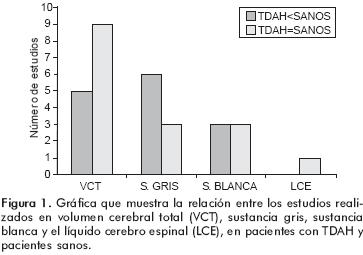
De la revisión de la literatura médica y del único meta–análisis publicado hasta la fecha, se concluye que a pesar de las diferencias metodológicas empleadas en los diferentes estudios y la inconsistencia de los hallazgos entre ellos, todo parece indicar que las estructuras anatómicas que cuentan con una mayor evidencia de ser menores en los sujetos con TDAH, en comparación con los controles sanos, son: los hemisferios cerebelosos, el vermis posterior e inferior, el esplenio del cuerpo calloso y, del hemisferio derecho, el cíngulo anterior y el cuerpo estriado (figura 1). Se han observado resultados de algunos estudios longitudinales,37,56,76,81 los cuales parecen indicar que estas alteraciones no se mantienen a lo largo del tiempo y, alrededor de la edad de 16 años, desaparecen las diferencias en volumen de las diversas estructuras anatómicas, lo que sugiere que se trata de un retraso de alrededor de dos años en el neurodesarrollo y probablemente del efecto de los cambios neuronales propios de la adolescencia como lo es la llamada poda sináptica.
Por otro lado, el 95% de los sujetos que han participado en los estudios de morfología han sido del sexo masculino y, principalmente, niños. Existen muy pocas publicaciones en las que se hayan incluido sujetos del sexo femenino y particularmente mujeres adultas con TDAH por lo que, teniendo en cuenta que el desarrollo del cerebro femenino es diferente al del masculino,89 se considera necesario realizar investigaciones que incluyan a sujetos del sexo femenino, adolescentes y adultos.
Factores como las metodologías y las secuencias utilizadas para la adquisición de las IRM, la resolución de los equipos de resonancia magnética, los métodos utilizados para medir las estructuras cerebrales, el uso de medicamentos previos en muchos de los sujetos que han participado en estos estudios y la presencia de comorbilidad, constituyen dificultades para obtener conclusiones definitivas sobre la anatomía patológica de este padecimiento y hacen que la misma siga siendo controversial. Desafortunadamente, la bibliografía está repleta de hallazgos inconsistentes en lo que se refiere a las alteraciones neuropsicológicas precisas y a las estructuras cerebrales implicadas. Los resultados heterogéneos de las muestras de niños estudiados pueden dar cuenta de estas inconsistencias, ya que las muestras varían mucho en cuanto al lugar de donde fueron extraídas (comunidad, clínicas pediátricas, clínicas psiquiátricas, etc.). Existen también datos heterogéneos en el tamaño de las muestras y en la selección de los criterios de inclusión y de exclusión. La misma diferencia clínica entre los sujetos con TDAH ha hecho sospechar a algunos investigadores que no existe un substrato anatómico exclusivo propio de este trastorno.90
El neurodesarrollo impacta negativamente en la consistencia de los hallazgos anatomopatológicos del TDAH, dado que el desarrollo de la corteza prefrontal es muy rápido y poco estable a lo largo de la infancia y produce una alta variabilidad intersujetos e intrasujetos.91 Por otro lado, los intentos por aclarar el substrato neuroanatómico y neuropsicológico del TDAH, eliminando lo más posible el efecto del neurodesarrollo por medio del estudio exclusivo de adultos, ha arrojado resultados también contradictorios; por ejemplo, estudios realizados con PET en adultos con TDAH han mostrado una disminución del metabolismo cerebral de la glucosa en la corteza prefrontal,92 pero este hallazgo no se reprodujo en adolescentes con TDAH.93 Además, el análisis de 33 publicaciones en las que se examinó el funcionamiento neuropsicológico de adultos con TDAH, indicó que las pruebas neuropsicológicas más frecuentemente utilizadas para evaluar la función ejecutiva, tales como el <<Wisconsin Card Sort Test>> y el puntaje de interferencia de la prueba <<Stroop>>, no pudieron discriminar entre sujetos adultos sanos y con TDAH.94 Estas inconsistencias despiertan dudas sobre el papel único de la corteza prefrontal y de sus conexiones como sustrato del TDAH. Por ello, algunos autores han sugerido la presencia de mecanismos alternativos en la fisiopatología del trastorno.95 Estos autores proponen que el daño anatómico en el TDAH probablemente se explique mejor por una alteración no cortical que permanece estable durante la infancia, por lo que el desarrollo de la corteza prefrontal y de sus sistemas relacionados compensan su alteración no cortical, ejerciendo un papel regulador <<Top–Down>> y con el consecuente cambio de la sintomatología a lo largo de la vida. De esta manera, la corteza prefrontal está íntimamente ligada a la sintomatología del TDAH, pero la causa de este trastorno no se localiza en ella, por lo que una alteración en la corteza prefrontal es necesaria pero no suficiente en un modelo de su fisiopatología. Es necesario considerar otros sistemas neurales para poder entenderla mejor.
Los ganglios de la base constan de tres núcleos que reciben aferencias (el núcleo caudado, el putamen y los núcleos subtalámicos) y dos núcleos que envían eferencias a diversas estructuras cerebrales (el globo pálido y el área 9 de la parte compacta de la sustancia negra mesencefálica). El núcleo caudado y el putamen reciben inervación glutaminérgica excitatoria que procede prácticamente de todas las regiones de la corteza cerebral y los núcleos subtalámicos reciben inervación de la corteza frontal motora (figura 2). Estas aferencias excitatorias activan un circuito que está formado por la corteza motora, el tálamo, los núcleos de la base, los núcleos subtalámicos y la sustancia negra.
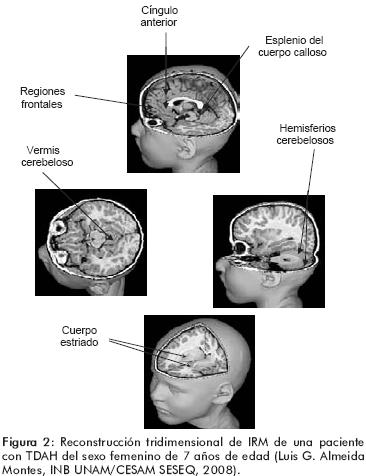
Las eferencias que inhiben o desinhiben al mesencéfalo y a los núcleos talámico–motores dependen de la actividad de este circuito. De esta manera, tal circuito sirve como nexo a través del cual las señales procedentes de las regiones corticales prefrontales, premotoras y motoras, inhiben programas motores que compiten entre sí y desinhiben aquellos que están listos para ser ejecutados.96 La eficiencia del funcionamiento de los núcleos basales depende de la inervación dopaminérgica procedente del mesencéfalo. Las neuronas dopaminérgicas del grupo celular A9 inervan profusamente al caudado y al putamen; esta inervación modula la excitación dopaminérgica procedente de la corteza en las neuronas espinosas estriatales por medio de mecanismos heterosinápticos, que incluyen la inhibición del receptor D2, facilitador de la liberación de glutamato, y la facilitación del receptor a glutamato NMDA, vía el receptor D1. El resultado neto de la inhibición dopaminérgica es disminuir las señales débiles y la actividad de fondo (<<noise>>, o ruido) y favorecer las señales glutaminérgicas intensas, incrementando la proporción señal–ruido (signal–noise ratio), lo que produce una conducta motora ordenada, oportuna y eficiente.97 Adicionalmente, las vías dopaminérgicas son muy sensibles a los agentes agresores durante el desarrollo cerebral en la infancia (por ejemplo, la anoxia) y las alteraciones producidas por tales eventos producen cambios a largo plazo en la actividad dopaminérgica.98 Por lo anterior es claro que los núcleos basales y las vías dopaminérgicas juegan un papel central en la fisiopatología del TDAH.
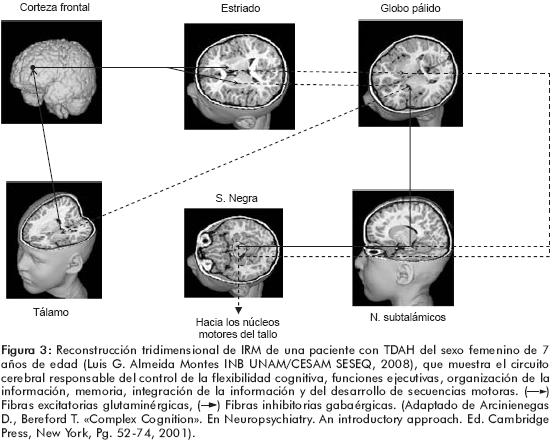
Algunos investigadores99 han propuesto que la respuesta lenta y variable que muestran en forma característica los sujetos con TDAH en las pruebas de vigilancia y de control inhibitorio reflejan una alteración en el alertamiento (disposición para recibir información), en la activación (disposición a responder a un estímulo) o en ambos. El alertamiento, junto con la orientación y la función ejecutiva, subyacen a la función cognitiva llamada atención.100 El origen de estas dificultades en sujetos con TDAH es el resultado de un pobre funcionamiento de las <<fuentes de energía>>. Esta hipótesis es conocida como el modelo cognitivo energético de Sanders del TDAH y, de acuerdo a él, en circunstancias normales la presencia de una señal estimula a la <<fuente de energía>> con el propósito de inducir la preparación motora para ejecutar una respuesta, así como de disminuir el tiempo de reacción de dicha respuesta. Más de medio siglo de investigación ha establecido que el locus coeruleus y otros núcleos noradrenérgicos del tallo cerebral son los sustratos del alertamiento y de la activación. El locus coeruleus está localizado en la región dorsal del puente que pertenece al grupo de neuronas noradrenérgicas A6 y desde aquí parten las eferencias que inervan a todo el neuroeje. La actividad tónica del locus coeruleus programa el estado conductual y, simultáneamente, los estímulos importantes producen disparos fásicos pronunciados de las neuronas del locus coeruleus. La noradrenalina modula la excitabilidad, la llegada de información, la proporción señal–ruido, la recepción y la dinámica temporal de las neuronas (corticales, talámicas, hipocámpicas y cerebelosas), por medio de mecanismos heterosinápticos que incluyen al receptor alfa 1 regulador de la liberación de glutamato y el potencial de membrana.101 El efecto neto es la reducción de la actividad espontánea y el incremento de las respuestas evocadas, lo que a su vez favorece el procesamiento y la respuesta hacia los estímulos sensoriales en el lóbulo frontal y mejora la cognición.102 De manera recíproca, la corteza frontal ejerce un papel modulador de la actividad del locus coeruleus. Este circuito es susceptible de sufrir daños durante las fases tempranas de la vida. Por estas razones, la fisiopatología del TDAH muy probablemente está relacionada con un funcionamiento deficiente de este circuito (figura 3).
El cerebelo está compuesto por dos hemisferios y el vermis. Se divide en tres lóbulos y 10 lobulillos discretos. Dentro del cerebelo se encuentran cuatro núcleos que están conectados, tanto de manera monosináptica como polisináptica, con virtualmente todo el neuroeje.103 Como se muestra en la figura 4, las proyecciones corticales hacen sinapsis con los núcleos pontinos de relevo y de ahí progresan por medio del pedúnculo cerebeloso medio para hacer sinapsis con las células de Purkinje y excitarlas de manera fásica. Las células de Purkinje inhiben la excitación tónica de los núcleos de salida del cerebelo (núcleos cerebelosos profundos), los cuales a su vez lanzan proyecciones hacia el tálamo, de donde parten proyecciones hacia la corteza frontal.104 De esta manera, el cerebelo y la corteza prefrontal, el cíngulo anterior y la corteza motora, están funcionalmente conectados. Además, el locus coeruleus y el área tegmental ventral (ATV) envían profusas proyecciones noradrenérgicas y dopaminérgicas que ejercen influencia sobre la actividad espontánea y evocada de las células de Purkinje105 y probablemente medien parte de los efectos terapéuticos de los medicamentos estimulantes en los sujetos con TDAH.106 Además de coordinar los movimientos, los resultados de diversos estudios han demostrado la participación del cerebelo en una serie de procesos cognitivos que incluyen la atención,107 el procesamiento temporal de la información108 y la anticipación de los eventos.109 Todos estos procesos cognitivos están alterados en sujetos con TDAH.110 (figura 5).


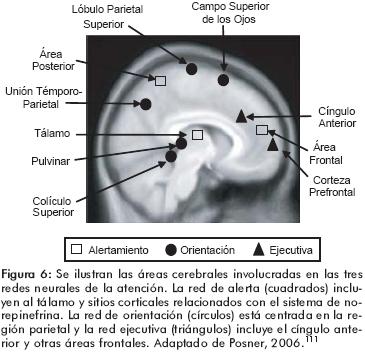
Además, existen otras áreas y estructuras encefálicas que participan en el proceso normal de la atención y que pueden estar involucradas en la fisiopatología del TDAH. Estas regiones incluyen al tálamo, la corteza parietal y a la unión temporoparietal, entre otras. Sin embargo, las mismas no han mostrado una clara diferencia en los estudios morfométricos realizados en los sujetos con TDAH. Los estudios de imágenes cerebrales realizados por el grupo de Fan y Posner sugieren la existencia de redes neuronales específicas que participan en la atención (figura 6). Existen al menos tres funciones de la atención cuyas redes neurales han sido demostradas por estudios de imágenes cerebrales: el alertamiento, el cual está involucrado en la adquisición y mantenimiento del estado de alerta; la orientación hacia un estímulo externo, y el control ejecutivo, el cual está involucrado con la resolución de conflictos entre sistemas neurales y que además regula los pensamientos y las emociones. La red cerebral que da cuenta del alertamiento está localizada en el sistema reticular ascendente, el tálamo y regiones parietales y frontales. La orientación tiene su sustrato anatómico en regiones tales como el colículo superior, el pulvinar, el lóbulo parietal y el campo ocular frontal. Finalmente, las funciones ejecutivas propias de la atención están vinculadas a redes neuronales que incluyen al cíngulo anterior y a la corteza prefrontal.111 Constituye una incógnita el por qué, en los estudios de imagen en el TDAH, algunas de estas estructuras no han sido consistentemente reportadas como más pequeñas en sujetos que lo padecen.
A pesar de los diseños longitudinales, y los estudios de casos y controles en los cuales se han separado por edad y sexo a los sujetos con TDAH con sus respectivos controles, la variable del neurodesarrollo aún puede explicar los hallazgos tan heterogéneos en lo que se refiere a su morfología. En un estudio recientemente publicado112 se muestran los factores heterogéneos del desarrollo cortical que están dados por la existencia de tres tipos de corteza en el cerebro humano: alocorteza, isocorteza y neocorteza. Los autores estudiaron a 375 sujetos con un rango de edad de 3.5 a 33 años, el 52% fueron masculinos y en el 94.6% se realizaron por lo menos dos estudios. Encontraron que la mayoría de la isocorteza de los lóbulos frontal lateral, temporal lateral, parietal y occipital tiene una trayectoria de desarrollo que es cúbica, la cual está caracterizada por un incremento en la infancia, un decremento en la adolescencia y una estabilización en la vida adulta. En contraste, un crecimiento caracterizado por un incremento inicial y un decremento terminal, en forma de <<U>> invertida (modelo cuadrático), se presenta en la ínsula y en el cíngulo anterior. Por último, se observa una trayectoria lineal en las regiones frontales orbitales posteriores y operculares, porciones de la corteza piriforme de la corteza temporal medial, las áreas del cíngulo que están debajo de la rodilla del cuerpo calloso y la corteza occípito–temporal medial. En general, estos autores encontraron una correspondencia entre el tipo de tejido y la forma de la trayectoria de desarrollo. Por ejemplo, en la parte más anterior y lateral del lóbulo frontal que se encuentra formada por isocorteza de seis capas, se presenta una curva de desarrollo cúbica. En contraste, la mayoría de la corteza orbital posterior presenta una trayectoria cuadrática y lineal. Estas regiones corresponden a la corteza transicional, la cual tiene menos capas y está menos desarrollada con la falta de la concentración típica de células no piramidales de la capa 4, la capa granular interna. En la parte más posterior de esta región se encuentra una trayectoria de desarrollo lineal y cuadrática, que caracteriza a la corteza piriforme, un tipo de alocorteza primitiva que es parte del sistema olfatorio. En la mayor parte del lóbulo frontal medial se sigue un desarrollo cúbico; el cíngulo anterior y medio presentan trayectorias cuadráticas y lineales. Se encuentran trayectorias cuadráticas en la ínsula, en regiones frontales inferiores laterales, en el temporal superior anterior y en su región polar; así como en el cíngulo anterior, cíngulo subcalloso y giro hipocampal y parahipocampal. Existen trayectorias de desarrollo lineales en el hipocampo, parahipocampo, giro fusiforme y lingual. Por último, estos autores determinaron la progresión del <<pico de grosor>> de la corteza cerebral. En la isocorteza, las áreas sensorio–motoras primarias alcanzan su pico de grosor antes que las áreas secundarias y polimodales adyacentes. En el lóbulo posterior, la primera área en alcanzar este pico es el área somato–sensorial (alrededor de los siete años), seguida por los polos occipitales (área visual primaria). En el lóbulo occipital derecho, el pico de grosor se encuentra alrededor de los siete años y en el izquierdo alrededor de los ocho. Las áreas polimodales parietales posteriores alcanzan su pico entre los nueve y 10 años. En el lóbulo frontal, el inicio del pico de grosor cortical se inicia en la corteza motora primaria y alcanza su máximo a la edad de nueve años, seguido de las áreas motoras suplementarias motoras a la edad de 10. La corteza dorsolateral prefrontal (CDLPF), a la cual se le atribuyen funciones cognitivas de <<mayor jerarquía>>, así como algunas áreas superiores del cíngulo, alcanzan su pico de grosor alrededor de los 10.5 años. En las áreas mediales, el pico del grosor de la corteza se alcanza primero en los polos frontales y occipitales y, de forma centrípeta, progresa hacia las regiones del cíngulo y de la corteza CDLPF. La progresión del desarrollo sigue una marcada tendencia dorso–ventral. Finalmente, existe una lateralidad bien definida en cada una de las regiones corticales: por ejemplo, en el cíngulo anterior derecho el pico del grosor cortical se alcanza a la edad de 13.8 años, mientras que en el izquierdo se alcanza a los 11.2. Es fácil comprender la variabilidad del desarrollo cerebral intra–sujetos e inter–sujetos, la cual probablemente no esté contemplada y controlada en los estudios morfológicos hechos en el TDAH. Esto puede ser una causa de las inconsistencias halladas en los estudios morfológicos.
Otra variable que puede influenciar el neurodesarrollo cerebral es el efecto genético. Por ejemplo, se ha postulado que el polimorfismo del gen que codifica a la enzima catecol–o–metiltransferasa, del trasportador de serotonina (5–HTTLPR), de la proteína G de membrana y del polimorfismo del receptor a dopamina D4 producen variaciones en el desarrollo del cerebro humano y pueden, en parte, explicar las inconsistencias encontradas en los estudios anatómicos realizados en el TDAH.
Por otro lado, el aprendizaje, la historia perinatal y la inteligencia por medio de factores neurotrópicos, influencian el desarrollo cerebral.37,112 Se ha encontrado, además, que el uso de estimulantes no afecta el desarrollo cerebral.56,113,114 El género también ejerce una influencia sobre el desarrollo cerebral: por ejemplo, el volumen cerebral total es alcanzado aproximadamente a la edad de 10.5 años en las niñas y a los 14.5 años en los niños. Tanto la sustancia gris cortical como la subcortical, siguen una trayectoria de desarrollo de <<U>> invertida que alcanza su tamaño máximo uno o dos años antes en mujeres que en varones. La sustancia gris se desarrolla hasta la edad de 24 años y los varones presentan un desarrollo más lento durante la adolescencia que las mujeres115. Además, la variabilidad del trastorno permanece en ocasiones incierta entre los géneros y esto es reflejado en la ausencia o inconsistencia de hallazgos en el sexo femenino respecto al masculino.114
Por último, es necesario considerar el papel influyente que puedan tener otros trastornos psiquiátricos en las estructuras cerebrales asociadas con el TDAH en la realización de los estudios de imagen.116
CONCLUSIÓN
Hasta ahora se empiezan a conocer cuáles son las estructuras anatómicas que muestran un volumen diferente entre sujetos sanos y con TDAH. Sin embargo, el conocimiento está aún incompleto y adolece de una falta de integración entre los hallazgos anatómico–funcionales en sujetos con TDAH y su correspondencia clínica, el impacto del desarrollo, el genotipo, la comorbilidad psiquiátrica, el sexo, etc. Por estas razones, hace falta la realización de estudios que correlacionen la disminución de volumen de las estructuras cerebrales con la gravedad del trastorno o los subtipos clínicos del TDAH, el uso de técnicas de medición de volúmenes cerebrales cada vez más precisas, la inclusión de sujetos del sexo femenino y en particular de los sujetos adultos, el uso de muestras de sujetos cuyas edades sean lo más homogéneas posible y la estratificación de las muestras de sujetos de acuerdo al genotipo.
REFERENCIAS
1. American Psychiatry Association. Diagnostic and Statistical Manual of Mental Disorders. Cuarta edición. Washington: American Psychiatric Publishing INC; 2000. [ Links ]
2. Biederman J. Attention–deficit/hyperactivity disorder: A selective overview. Biol Psychiatry 2005;57:1215–1220. [ Links ]
3. Kessler R, Adler L, Barkley R, Biederman J, Conners C et al. The prevalence and correlates of adult ADHD in the United States. Results of national comorbility survey replication. Am J Psychiatry 2006;163:716–723. [ Links ]
4. Fayyad J, De Graff R, Kessler R, Alonso J, Angermeyer M et al. Cross–national prevalence and correlates of adult attention–deficit/hyperactivity disorder. Br J Psychiatry 2007;190:402–409. [ Links ]
5. Almeida L, Hernández A, Ricardo–Garcell J. ADHD prevalence in adult outpatients with nonpsychotic psychiatric illnesses. J Atten Disord 2007;11:150–156. [ Links ]
6. Biederman J, Mick E, Faraone S. Age–dependent decline of symptoms of attention–deficit/hyperactivity disorder: Impact of remission definition and symptom type. Am J Psychiatry 2000;157:816–818. [ Links ]
7. Helgeland M, Kjelsberg E, Torgersen S. Continuities between emotional and disruptive behavior disorders in adolescence and personality disorders in adulthood. Am J Psychiatry 2005;162:1941–1947. [ Links ]
8.Mannuzza S, Klein R, Bessler A, Malloy P, La Padula M. Adult psychiatric status of hyperactive boys grown up. Am J Psychiatry 1998;155:493–498. [ Links ]
9. Dalsgaard S, Mortensen P, Frydenberg M, Thomsen P. Conduct problems, gender and adult psychiatric outcome of children with attention–deficit/hyperactivity disorder. Br J Psychiatry 2002;181:416–421. [ Links ]
10. Rosler M, Retz W, Retz–Junginger P, Hengesch G, Schneider M et al. Prevalence of attention–deficit/hyperactivity disorder (ADHD) and co–morbid disorders in young male prison inmates. Eur Arch Psychiatry Clin Neurosci 2004;254:365–371. [ Links ]
11. Nierenberg A, Miyahara S, Spencer T, Wisniewski S, Otto M et al. Clinical and diagnostic implications of lifetime attention–deficit/hyperactivity disorder comorbidity in adults with bipolar disorder: Data from the first 1000 STEP–BD participants. Biol Psychiatry 2005;57:1467–1473. [ Links ]
12. Fischer M, Barkley R, Smallish L, Fletcher K. Hyperactive children as young adults: Driving abilities, safe driving behavior, and adverse driving outcomes. Accid Anal Prev 2007;39:94–105. [ Links ]
13. Faraone S, Biederman J, Chen W, Krifcher B, Keenan K et al. Segregation analysis of attention–deficit/hyperactivity disorder: Evidence for single gene transmission. Psychiatr Genet 1992;2:257–275. [ Links ]
14. Faraone S, Perlis R, Doyle A, Smoller J, Gorlanick J et al. Molecular genetics of attention–deficit/hyperactivity disorder. Biol Psychiatry 2005;57:1313–1323. [ Links ]
15. Sprich–Buckminster S, Crawford M, Mundy E, Faraone S. Adoptive and biological families of children and adolescents with ADHD. J Am Acad Child Adolesc Psychiatry 2000;39:1432–1437. [ Links ]
16. Faraone S, Doyle A, Mick E, Biederman J. Meta–analysis of the Association Between the 7–Repeat Allele of the Dopamine D(4) Receptor Gene and Attention–Deficit/Hyperactivity Disorder. Am J Psychiatry 2001;158:1052–1057. [ Links ]
17. Kupfer D, First M, Regier D. A Research agenda for DSM–V. First Edition. Washington DC. Am Psychiatr Assoc; 2002. [ Links ]
18. Lynam D, Caspi A, Moffitt T. Longitudinal evidence that psychopathology scorers in early adolescence predict adult psychopathology. J Ab–norm Psychol 2007;116:115–165. [ Links ]
19. Porges SW. Asserting the role of biobehavioral sciences in translational research: The behavioral neurobiology revolution. Dev Psychopathol 2006;18:923–933. [ Links ]
20. Coghill D, Nigg J, Rothemberg A. Whither casual models in the neuroscience of ADHD? Dev Science 2005;8:105–114. [ Links ]
21. Volkmar F. Toward understanding the basis of ADHD. Am J Psychiatry 2005;162:1043–1044. [ Links ]
22. Spencer T, Biederman J, Wilens T. Overview and neurobiology of attention–deficit/hyperactivity disorder. J Clin Psychiatry 2002;63:3–9. [ Links ]
23. Biederman J, Faraone S, Keenan K. Family–genetic and psychosocial risk factors in DSM III attention–deficit/hyperactivity disorder. J Acad Child Adolesc Psychiatry 1990;29:526–533. [ Links ]
24. Banerejee T, Middleton F, Faraone S. Environmental risk factors for attention–deficit/hyperactivity disorders. Acta Pediatr 2007;96:1269–1274. [ Links ]
25. Linnet K, Dalsgaard S, Obel C. Maternal lifestyle factors in pregnancy risk of attention–deficit/hyperactivity disorder and associated behaviors: Review of the current evidence. Am J Psychiatry 2003;160:1028–1040. [ Links ]
26. Thapar A, Holmes J, Poulton K. Genetic basis of attention–deficit and hyperactivity. Br J Psychiatry 1999;174:105–111. [ Links ]
27. Langley K, Rice F, Van den Bree M. Maternal smoking during pregnancy as an environmental risk factor for attention–deficit/hyperactivity disorder behaviors. A review. Minerva Pediatr 2005;57:359–351. [ Links ]
28. Smalley SL. Genetic influences in childhood–onset psychiatric disorders: Autism and attention–deficit/hyperactivity disorder. Am J Hum Genet 1997;60:1276–1282. [ Links ]
29. Sthal SM. Attention–deficit/hyperactivity disorder and its treatment. En: Sthal's essential psychopharmacology. neuroscientific basis and practical applications. New York: Cambridge University Press; 2008; pp. 863–898. [ Links ]
30. Botting N, Powls A, Cooke R. Attention–deficit/hyperactivity disorders and other psychiatric outcomes in very low birth weight children at 12 years. J Psychol Psychiatr 2007;38:931–941. [ Links ]
31. Serrano P, Friederichsen A, Piña S, García F, Almeida L et al. Frecuencia de complicaciones perinatales en pacientes con trastorno por déficit de la atención e hiperactividad, otras enfermedades psiquiátricas infantiles y sujetos sanos. Psiq Biol 2003;10:183–188. [ Links ]
32. Kahn R, Khoury J, Nichols W. Role of dopamine transporter genotype and maternal parental smoking in childhood hyperactive–impulsive, inattentive, and oppositional behaviors. J Pediatr 2003;143:104–110. [ Links ]
33. Laucht M, Skowronek M, Becker K. Interacting effects of the dopamine transporter gene and psychosocial adversity on attention–deficit/hyperactivity disorder symptoms among 15–year olds from high risk community sample. Arch Gen Psychiatry 2007;64:585–90. [ Links ]
34. Levitt P. Developmental neurobiology and clinical disorders: Lost in translation? Neuron 2005;46:407–412. [ Links ]
35. Karlin A, Castellanos X. Brain development and ADHD. Clin Psychol Rev 2006;26:433–444. [ Links ]
36. Kieling C, Goncalves R, Tannock R, Castellanos X. Neurobiology of attention–deficit/hyperactivity disorder. Child Adolesc Psychiatr Clin N Am 2008;17:285–307. [ Links ]
37. Shaw P, Gornick M, Lerch J, Addington A, Seal J et al. Polymorphisms of the dopamine D4 receptor, clinical outcome, and cortical structure in attention–deficit/hyperactivity disorder. Arch Gen Psychiatry 2007;64:921–931. [ Links ]
38. Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch J et al. Attention– deficit/hyperactivity disorder is characterized by a delay in cortical maturation. PANS 2007;104:19646–19654. [ Links ]
39. Shaywitz B, Yager R, Klopper J. Selective brain dopamine depletion in developing rats: An experimental model of minimal brain dysfunction. Science 1976;191:305–308. [ Links ]
40. Raskin L, Shaywitz B, Anderson G. Differential Effects of selective dopamine, norepinephrine or catecholamine depletion on activity and learning in the developing rat. Pharmacol Biochem Behav 1983;19:743–749. [ Links ]
41. Dougherty D, Bonab A, Spencer T. Dopamine transporter density in patients with attention–deficit/hyperactivity disorder. Lancet 1999;354:2132–2133. [ Links ]
42. Krause K, Dresel S, Krause J. Increased stratial dopamine transporter in adult patients with attention–deficit/hyperactivity disorder: Effects of methylphenidate as measured by single photon emission tomography. Neurosci Lett 2000;285:107–110. [ Links ]
43. Ernst M, Zametkin A, Matochick J. DOPA decarboxylase activity in attention–deficit/hyperactivity disorder adults. A [Flourine–18] fluorodopa positron emission tomographic study. J Neurosci 1998;18:5901–5907. [ Links ]
44. Volkow N, Wang G, Fowler J. Imaging the effects of methylphenidate on brain dopamine: New model on its therapeutic actions for attention–deficit/hyperactivity disorder. Biol Psychiatry 2005;57:1410–1415. [ Links ]
45. Pliszka S. The neuropharmacology of attention–deficit/hyperactivity disorder. Biol Psychiatry 2005;57:1385–1390. [ Links ]
46. Faraone S, Biederman J. Neurobiology of attention–deficit/hyperactivity disorder. En: Neurobiology of mental illness. Segunda edición. Charney D, Nestler E (eds.). New York: Oxford University Press; 2004; pg. 979–1006. [ Links ]
47. Fan J, McCadliss B, Fosella J, Flombaum J, Posner M. The Activation of attentional networks. Neuroimage 2005;26:471–479. [ Links ]
48. Almeida L. Alteraciones anatómico–funcionales en el trastorno por déficit de la atención con hiperactividad. Salud Mental 2005;28:1–12. [ Links ]
49. Haacke W. Magnetic resonance imaging: A preview. En: Magnetic resonance imaging. Physical principles & sequence design. Thompson M, Venkatesan R (eds.) Wiley–Lis., New York: 1999; pp. 1–13. [ Links ]
50. Aylward E, Reiss A, Reader M, Singer H, Brown J et al. Basal ganglia volumes in children with attention–deficit/hyperactivity disorder. J Child Neurol 1996;11:112–115. [ Links ]
51. Baumgardner T, Singer H, Denckla M, Rubin M, Abrams M et al. Corpus callosum morphology in children with Tourette Syndrome and attention–deficit/hyperactivity disorder. Neurology 1996;47:1–6. [ Links ]
52. Berquin P, Giedd J, Jacobsen L, Hamburger S, Krain A et al. Cerebellum in attention–deficit/hyperactivity disorder: A morphometric MRI study. Neurology 1998;50:1087–1093. [ Links ]
53. Bussing R, Grudnik J, Mason D, Wasiak M, Leonard C. ADHD and conduct disorder: An MRI study in a community sample. World J Biol Psychiatry 2002;3:216–220. [ Links ]
54. Castellanos F, Giedd J, Marsh W, Hamburger S, Vaituzis A et al. Quantitative brain magnetic resonance imaging in attention–deûcit/hyperactivity disorder. Arch Gene Psychiatry 1996;53:607– 616. [ Links ]
55. Castellanos F, Giedd J, Berquin P, Walter J, Sharp W et al. Quantitative brain magnetic resonance imaging in girls with attention–deficit/hyperactivity disorder. Arch Gene Psychiatry 2001;58:289–295. [ Links ]
56. Castellanos F, Lee P, Sharp W, Jeffries N, Greenstein D et al. Developmental trajectories of brain volume abnormalities in children and adolescents with attention–deficit/hyperactivity disorder. JAMA 2002;288: 1740–1748. [ Links ]
57. Durston S, Hulshoff Pol H, Schnack H, Buitelaar J, Steenhuis M et al. Magnetic resonance imaging of boys with attention–deûcit/hyperactivity disorder and their unaffected siblings. J Am Acad Child Adolesc Psychiatry 2004;43:332–340. [ Links ]
58. Filipek P, Semrud–Clikeman M, Steingard R, Renshaw P, Kennedy D et al. Volumetric MRI analysis: Comparing subjects having attention–deûcit/ hyperactivity disorder with normal controls. Neurology 1997;48:589–601. [ Links ]
59. Giedd J, Castellanos F, Casey B, Kozuch P, King A et al. Quantitative morphology of the corpus callosum in attention–deficit/hyperactivity disorder. Am J Psychiatry 1994;151:665–669. [ Links ]
60. Hill D, Yeo R, Campbell R, Hart B, Vigil J et al. Magnetic resonance imaging correlates of attention–deficit/hyperactivity disorder in children. Neuropsychology 2003;17:496–506. [ Links ]
61. Hynd G, Semrud–Clikeman M, Lorys A, Novey E, Eliopulos D. Brain morphology in developmental dyslexia and attention deficit disorder/ hyperactivity. Arch Neurol 1990;47:919–926. [ Links ]
62. Hynd G, Semrud–Clikeman M, Lorys A, Novey E, Eliopulos D et al. Corpus callosum morphology in attention–deficit hyperactivity disorder: Morphometric analysis of MRI. J Learn Disabil 1991;24:141–146. [ Links ]
63. Hynd G, Hern K, Novey E, Eliopulos D, Marshall R et al. Attention–deficit/hyperactivity disorder and asymmetry of the caudate nucleus. J Child Neurol 1993;8:339–347. [ Links ]
64. Kates W, Frederikse M, Mostofsky S, Folley B, Cooper K et al. MRI parcellation of the frontal lobe in boys with attention–deficit/hyperactivity disorder or Tourette syndrome. Psychiatry Research Neuroimaging 2002;116:63–81. [ Links ]
65. Lyoo I, Noam G, Lee C, Lee H, Kennedy B et al. The corpus callosum and cateral ventricles in children with attention–deficit/hyperactivity disorder: A brain magnetic resonance imaging study. Biol Psychiatry 1996;40:1060–1063. [ Links ]
66. Mataro M, García–Sánchez C, Junque C, Estevez–Gonzalez A, Pujol J. Magnetic resonance imaging measurement of the caudate nucleus in adolescents with attention–deficit/hyperactivity disorder and its relationship with neuropsychological and behavioral measures. Arch Neurol 1997;54:963–968. [ Links ]
67. Mostofsky S, Reiss A, Lockhart P, Denckla M. Evaluation of cerebellar size in attention–deûcit/hyperactivity disorder. J Child Neurol 1998;13:434–439. [ Links ]
68. Mostofsky S, Cooper L, Kates W, Dencka M, Kaufmann W. Smaller prefrontal and premotor volumes in boys with attention–deficit/hyperactivity disorder. Biol Psychiatry 2002;52:785–794. [ Links ]
69. Pineda D, Restrepo M, Sarmiento R, Gutierrez J, Vargas S et al. Statistical analyses of structural magnetic resonance imaging of the head of the caudate nucleus in Colombian children with attention–deficit/hyperactivity disorder. J Child Neurol 2002;17:97–105. [ Links ]
70. Semrud–Clikeman M, Filipek P, Biederman J, Steingard R, Kennedy D et al. Attention–deficit/hyperactivity disorder: magnetic resonance imaging morphometric analysis of the corpus callosum. J Am Acad Child Adolesc Psychiatry 1994;33:875–881. [ Links ]
71. Seidman L, Valera E, Makris N, Monuteaux M, Boriel D et al. Dorsolateral prefrontal and anterior cingulated cortex volumetric abnormalities in adults with attention–deficit/hyperactivity disorder. Biol Psychiatry 2006;60:1071–1080. [ Links ]
72. Biederman J, Makris N, Valera E, Monuteaux M, Goldstein J et al. Towards further understanding of the co–morbidity between attention–deficit/hyperactivity disorder and bipolar disorder: a MRI study of brain volumes. Psychol Med 2008;38:1045–1056. [ Links ]
73. Carmona S, Vilarroya O, Bielsa A, Tremols V, Rovira M, Tomas J et al. Global and regional gray matter reductions in ADHD: A voxel–based morphometric study. Neurosci Lett 2005;389:88–93. [ Links ]
74. Castellanos F, Sharp W, Gottesman R, Greenstein D, Giedd J et al. Anatomic brain abnormalities in monozygotic twins discordant for attention–deficit/hyperactivity disorder. Am J Psychiatry 2003;160:1693–1696. [ Links ]
75. Hesslinger B, Tebartz van Elst, Thiel T, Heagele K, Hennig J et al. Frontoorbital volume reductions in adult patients with attention–deficit/hyperactivity disorder. Neurosci Lett 2002;328:319–321. [ Links ]
76. Mackie S, Shaw P, Lenroot R, Pierson R, Greenstein D et al. Cerebellar development and clinical outcome in attention–deficit/hyperactivity disorder. Am J Psychiatry 2007;164:647–655. [ Links ]
77. Makris N, Biederman J, Valera E, Bush G, Kaiser J et al. Cortical thinning of the attention and executive function networks in adults with attention–deficit/hyperactivity disorder. Cerebral Cortex 2007; 17:1364–1375. [ Links ]
78. McAlonan G, Cheung V, Cheung C, Chua S, Murphy D et al. Mapping brain structure in attention deficit–hyperactivity disorder: A voxel–based MRI study of regional grey and white matter volume. Psychiatry Research Neuroimaging 2007;154:171–180. [ Links ]
79. Overmeyer S, Bullmore E, Suckling J, Simmons A, Wiliams S et al. Distributed grey and white matter deficits in hyperkinetic disorder: MRI evidence for anatomical abnormality in an attentional network. Psychol Med 2001;31:1425–1435. [ Links ]
80. Pliszka S, Lancaster J, Liotti M, Semrud–Clikeman M. Volumetric MRI differences in treatment–naïve versus chronically treated children with ADHD. Neurology 2006;67:1023–1027. [ Links ]
81. Shaw P, Lerch J, Greensteien D, Sharp W, Clasen L et al. Longitudinal thickness and clinical outcome in children and adolescents with atten–tion–deficit/hyperactivity disorder. Arch Gene Psychiatry 2006;63:540–549. [ Links ]
82. Sowell E, Thompson P, Welcome S, Henkenius A, Toga A et al. Cortical abnormalities in children and adolescents with attention–deficit/hyperactivity disorder. Lancet 2003;362:1699–1707. [ Links ]
83. Tremols V, Bielsa A, Soliva J, Raheb C, Carmona S et al. Differential abnormalities of the head and body of the caudate nucleus in attention–deficit–/hyperactivity disorder. Psychiatry Research Neuroimaging 2008;163:270–278. [ Links ]
84. Wellington T, Semrud–Clikeman M, Gregory A, Murphy J, Lancaster J. Magnetic resonance imaging volumetric analysis of the putamen in children with ADHD: Combined type versus control. J Atten Disord 2006;10:171–180. [ Links ]
85. Glass GV. Integrating findings: The meta–analysis of research. En: Review of research in education. Shulman L (ed). Peacock; 1977; pp. 351–379. [ Links ]
86. Cohen J. Statistical power analysis for the behavioral sciences. Segunda edición. Academic Press; 1988. [ Links ]
87. Valera E. Advances in neuroimaging and genetics in ADHD. Presentado en el simposio APA 2008 Annual Meeting. Washington DC, mayo, 2008. [ Links ]
88. Valera E, Faraone S, Murray K, Seidman L. Meta–analysis of structural imaging findings in attention–deficit/hyperactivity disorder. Biol Psychiatry 2007;61:1361–1369. [ Links ]
89. Rhoshel L, Nitin G, Deanna G, Molloy E, Wallace G et al. Sexual dimorphism of brain developmental trajectories during childhood and adolescence. Neuroimage 2007;36:1065–1073. [ Links ]
90. Pennington B. Toward a new model of neuropshycological model of attention–deficit/hyperactivity disorder: Subtypes and multiple deficits. Biol Psychiatry 2005;67:1221–1223. [ Links ]
91. Barkovich A. Magnetic resonance techniques in the assessment of myelin and myelination. J Inherit Metab Disord 2005;28:311–343. [ Links ]
92. Zametkin A, Nordahl T, Gross M, Kings A, Semple WE et al. Cerebral glucose metabolism in adults with hyperactivity of childhood onset. N Engl J Med 1990;323:1361–1366. [ Links ]
93. Ernst M, Cohen R, Liebenauer L, Jons R, Zametkin A. Cerebral glucose metabolism in adolescent girls with attention–deficit/hyperactivity disorder. J Acad Child Adolesc Psychiatry 1997;36:1399–1406. [ Links ]
94. Hervey A, Epstein J, Curry J. Neuropsychology of adults with attention–deficit/hyperactivity disorder: A meta–analytic review. Neuropsychology 2004;18:485–503. [ Links ]
95. Halperin J, Schulz K. Revisiting the role of the prefrontal cortex in the pathophysiology of attention–deficit/hyperactivity disorder. Psychol Bull 2006;132:560–581. [ Links ]
96. Mink W. The basal ganglia. Focused selection and inhibition of competing motor programs. Prog Neurobiol 1996;50:381–425. [ Links ]
97. Volkow N, Wang G, Fowler J, Gatley S, Logan J et al. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry 1998;155:1325–1331. [ Links ]
98. Chen Y, Engidawork E, Loidl F, Anna D, Goiny M et al. Short and long term effects of perinatal asphyxia on monoamine, aminoacid and glycolysis product levels measured in the basal ganglia of the rat. Dev Brain Res 1997;104:10–30. [ Links ]
99. Sergeant A. The cognitive–energetic model: An empirical approach to attention–deficit/hyperactivity disorder. Neurosci Biobehav Rev 2000;24:7–12. [ Links ]
100. Posner I, Petersen E. The attention system of the human brain. Annu Rev Neurosci 1990;13:25–42. [ Links ]
101. Marek J, Aghajanian K. 5HT2A receptor or alpha 1 adrenoreceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur J Pharmacol 1999;367:197–206. [ Links ]
102. Arnsten A, Steere J, Hunt D. The contribution of Alpha 2–noradrenergic mechanisms to prefrontal cortical cognitive function. Potential significance for attention–deficit/hyperactivity disorder. Arch Gene Psychiatry 1996;53:448–455. [ Links ]
103. Middleton F, Strick L. Basal ganglia and cerebellar loops. Motor and cognitive circuit. Brain Res Rev 2000;31:236–250. [ Links ]
104. Shinoda Y, Sugihara I, Wu S, Sugiuchi Y. The entire trajectory of single climbing and mossy fibers in the cerebellar nuclei and cortex. Prog Brain Res 2000;124:173–186. [ Links ]
105. Schweighofer N, Doya K, Kuroda S. Cerebellar aminergic neuromodulation: Towards a functional understanding. Brain Res Rev 2004;44:103–116. [ Links ]
106. Anderson M, Polcari A, Lowen B, Renshaw F, Teicher M. Effects of methylphenidate on functional magnetic resonance relaxometry of the cerebellar vermis in boys with ADHD. Am J Psychiatry 2002;159:1322–1328. [ Links ]
107. Allen G, Buxton B, Wong E, Courchesne E. Attentional activation of the cerebellum independent of motor involvement. Sicence 1997; 275:1940–1943. [ Links ]
108. Ivry B, Keele W. Timing functions of the cerebellum. J Cognitive Neuroscience 1989;1:136–152. [ Links ]
109. Tesche C, Karhu J. Anticipatory cerebellar responses during somato–sensory omission in man. Hum Brain Mapp 2000;9:119–142. [ Links ]
110. Smith A, Taylor E, Rogers W, Newman S, Rubia K. Evidence for a pure time perception deficit in children with ADHD. J Child Psychol Psychiatry 2002;43:529–542. [ Links ]
111. Posner I, Sheese B, Odlulas Y, Tang Y. Analyzing and shaping human attentional networks. Neural Netw 2006;19:1422–1429. [ Links ]
112. Shaw P, Noor J, Lerch J, Eckstrand K, Lenroot R et al. Neurodevelompental trajectories of the human cerebral cortex. J Neuroscience 2008;28:3586–3594. [ Links ]
113. Shaw P, Sharp W, Morrison M, Eckstrand K, Greentein D et al. Psychostimulant treatment and the developing cortex in attention–deficit/hyperactivity disorder. Am J Psychiatric 2009;166:58–63. [ Links ]
114. Qiu A, Crocetti D, Adler M, Mahone M, Denckla M et al. Basal ganglia volume and shape in children with attention–deficit/hyperactivity disorder. Am J Psychiatry 2009;166:74–82. [ Links ]
115. Lenroot R, Gogtay N, Greenstein D, Wells E, Wallace G Sexual dimorphism of brian developmental trajectories during childhood and adolescence. Neuroimage 2007;36:1065–1073. [ Links ]
116. Luders E, Narr K, Hamilton L, Phillips O, Thompson P et al. Decreased callosal thickness in attention–deûcit/hyperactivity disorder. Biol Psychiatry 2009;65:84–88. [ Links ]
* Los resúmenes en inglés y español aparecieron en la primera parte de este artículo. Salud Mental, Vol. 32, No. 6, 2009.

