










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista mexicana de fitopatología
versão On-line ISSN 2007-8080versão impressa ISSN 0185-3309
Rev. mex. fitopatol vol.35 no.3 Texcoco Set. 2017
https://doi.org/10.18781/r.mex.fit.1706-1
Review articles
Evolution of diagnostic technics for plant viruses
1Biociencia, S. A. de C. V.; Agustín Melgar 2317, Col. Reforma, C.P. 64550, Monterrey, N. L.
The symptoms caused by viruses in diseased plants were the first way to detect and identify the viruses that affected them and named them according to the symptoms they produced. The use of differential plants infected by mechanical transmission, by grafting or vectors, increased the ability to detect and identify many of the phytopathogenic viruses and also led to confounding other diseases caused by other infectious agents with viruses. Serology detection uses the interaction of the viral protein as an antigen against the antibodies produced against them by a vertebrate. The first serological methods used for viral detection were antigen-antibody precipitation in liquid medium, followed by agar double diffusion and finally by enzyme-linked immunosorbent assay, very economical in the use of reagents, very sensitive and uses a Solid medium for antigen or antibody immobilization which may be different plastics or membranes. Detection of viral nucleic acid by nucleic acid hybridization or polymerase chain reaction is more sensitive than serology, and the latter method combined with second generation sequencers (Next Generation Sequencing) have revolutionized detection of viruses in plants.
Key words: Detection of viruses; differential plants; serology; ELISA; nucleic acid hybridization; PCR; NGS
La sintomatología producida por los virus en las plantas enfermas fue la primera forma de detectar e identificar los virus que las afectaban y los nombraron de acuerdo a la sintomatología que producían. El uso de plantas diferenciales infectadas mediante transmisión mecánica, por injerto o vectores, amplió la capacidad de detectar e identificar muchos de los virus fitopatógenos y también llevó a confundir con virosis otras enfermedades causadas por otros agentes infecciosos. La detección por serología usa la interacción de la proteína viral como antígeno contra los anticuerpos producidos contra ellos por un vertebrado. Los primeros métodos serológicos usados para la detección viral fueron de precipitación antígenos-anticuerpo en medio líquido, seguido por la doble difusión en agar y finalmente por el ensayo de inmunoabsorción ligado a enzima, muy económico en el uso de reactivos, muy sensible y usa un medio sólido de inmovilización del antígeno o del anticuerpo que pueden ser diferentes plásticos o membranas. La detección del ácido nucleico viral mediante el método de hibridación de ácidos nucleicos o por la reacción en cadena de la polimerasa es más sensible que la serología y éste último método combinado con los secuenciadores de la segunda generación (Next Generation Sequencing) han revolucionado la detección de virus en los vegetales.
Palabras clave: Detección de virus; plantas diferenciales; serología; ELISA; hibridación de ácidos nucleicos; PCR; NGS
INTRODUCTION
Unlike other plant pathogens, the management of viral diseases based on direct methods for their control, such as the use of virucides, have not been developed to date. Therefore, viral diseases are fought with indirect strategies, such as fighting the vector insect, eliminating diseased plants, or avoiding the planting of infected seeds. Because of this, the methods to detect and identify viruses are crucial for handling viral diseases. Detection methods must be as convenient, effective, specific, and quick as possible (Joo-jin J. 2014) and are acknowledged as the basic instrument of virology, because the accuracy of the studies depend directly on the sensitivity of these techniques (Salazar 1995).
Viruses have coevolved for millions of years, hosting the three domains of life forms, achaebacteria, Bacteria, and Eukaryotes (Gergerich and Dolja 2006); however, their knowledge is very recent. Studies on viruses begins with the works by Mayer in 1886, who mechanically transmitted the Tobacco Mosaic Virus (TMV) from a diseased plant to a healthy plant; Iwanowski, in 1892, proved that the sap of these plants remained ineffective after being bassed through a porcelain bacteria-proof filter, and Beijerinck, in 1898, discovered the causal agent as “Contagium vivum fluidum”, and first introduced the word “virus” to differentiate it from the contaigous corpuscular fuid containing bacteria or fungal spores, thus giving rise to the science of Virology (Hull R. 2014). Ever since then, researchers have searched for a way to study viruses to detect them in their hosts, identify, and classify all new viruses that were being discovered in this emerging branch of science.
Detection by symptoms
The symptoms produced by the viruses in the plants was the first way to detect and identify viruses in the late 19th Century, and their names were related to the symptoms it produced, such as the Tobacco mosaic virus (TMV), the Papaya ringspot virus (PRSV), etc. However, scientists soon realized that many variants of the same virus produced very different symptoms, and on the other hand, many different viruses produced very similar symptoms, and in addition, plants also presented symptoms similar to a virosis as a response to unvaforable weather conditions, nutient balances in soil minerals, infections by non-viral pathogens, damage caused by insects, mites, or nematodes. Although the symptmos provided crucial information on viral diseases, much field experience was required to make decisions based only on symptoms to identify a virus. Generally, field inspections must be accompanied by other tests for the correct diagnosis of a viral infection (Naidú and Hughes. 2001).
During the period between 1900 and 1935, there was more attention on the description and identification by the symptoms of the disease, both macroscopically and by abnormalities caused inside the cells observed under light microscopes, called viral inclusions. These were the only aspects that could be studied with the techniques at hand. All was confusing, and the only fact scientists had about viruses was that they were very small and caused diseases. In 1929, Holmes showed that local lesions produced by mechanical inoculation in certain hosts could be used to count the amount of infective viruses. This technique helped study come of the properties of viruses, such as the point of thermal inactivation, point of maximum dilution, and in vitro longevity, and it paved the way for the future isolation and purification of the viruses.
Using the mechanical transmission technique, many of the plant diseases caused by filterable viruses were described between 1900 and 1935, although it was soon found that many diseases with symptoms similar to viral diseases were not mechanically transmitted and the filterability criterion could not be applied. However, its infectious nature could be proven by transmission by grafting or by vectors. In this way, many of the diseases were caused by viruses that were not mechancially transmitted, but many others, with symptoms of yellowing or witch’s broom, that were attributed to viruses, although it was later proven that they were caused by phytoplasms or Spiroplasms or other non-cultivable bacteria and not mechanically transmissible.
Detection with differentiated plants
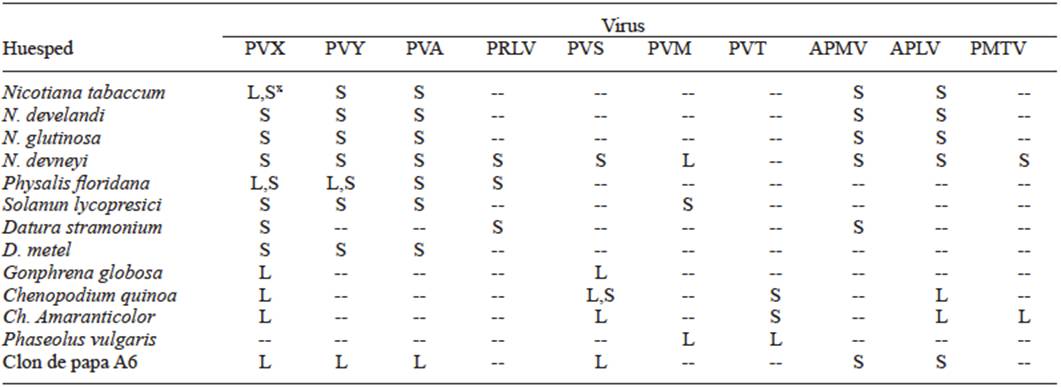
In 1931, Smith worked with viruses on potatoes and proved that the symptoms could be caused by the combination of several viruses, and in this way, by using differential plants and transmission by insects, he was able to separate Potato virus X (PVX) from Potato virus Y (PVY). By transmitting the latter with the aphid Myzus persicae that did not transmit PVX and by mechanical inoculation in Datura stramonium he separated PVX, since this plant is immune to virus Y. In this way, he was able to work with pure viruses to determine their host ranges, and for the first time, was able to observe the differences in the virulence of the isolations of one same virus due to the differences in symptoms produced. Until that moment, the use of differential plants and their range of hosts, using mechanical inoculation or by grafting o with the use of vectors, was the only way to identify viruses in plants. Tables 1, 2, and 3 show the different symptoms in the differential plants that were used to identify the Papaya ringspot virus (PRSV), for the Cucumber Mosaic Virus (CMV) (Kelaniyangoda and Madhubashini 2008), and 10 different viruses in potatoes (Salazar 1995). In this way, symptom comparison charts were drawn so all viruses could be identified, using mechanical transmission or by vectors or grafting. The use of differential plants to detect viruses is still common in several fruit trees such as citrus, pome, and rosaceae plants (Roistacher 1991).
Table 1 Range of hosts and symptoms for the diagnosis of the PRSV (three weeks postinoculation).
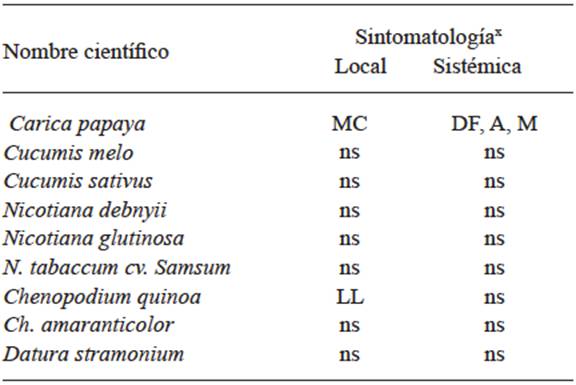
x MC= Chlorotic spot; DF= Foliar distortion; A= Stunting; M= Mosaic; LL= Local lesions; ns= No symptoms.
Table 2 Range of hosts and symptoms for the diagnosis of CMV.
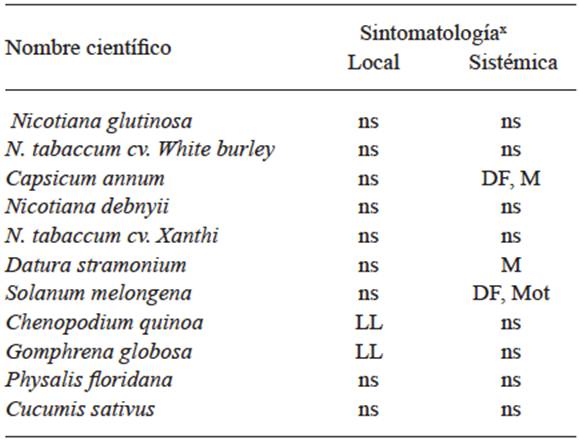
x DF= Foliar distortion; MC= Chlorotic spots; Mot= Dotted; M= Mosaic; LL= Local lesions; ns= No symptoms.
Detection by serology
The birth of serology for the identification of viruses that affect plants began with the discovery by Beale in 1928, who showed that the plants infected by the Tobacco mosaic virus (TMV) contained a specific antigen, different to those of the healthy plant, when both saps were injected into different rabbits. Later, in 1933, Gratia showed that plants infected with different viruses contained different specific antigens, and finally, Chester, in 1935 and 1936, showed that the different TMV and PVX isolations could be told apart serologically (Hull R 2004).
As progress was being made in the methodology for the purification of viruses, mainly with the discovery by Brakke (1951 and 1953) of centrifugation in differential gradients using different concentrations of lithium chloride, caesium chloride ot sulphate, and later, with the development of centrifugation in sacarose gradients of 10 to 40%, on which the infective sap was formed, (Lister, 1966 and 1968), the virus was separate, forming a strip, in which its density would equal the density of the sacarose. With the purified viruses, the specific antisera were formed for each of the purified viruses that already helped for the differential identification of the different viruses that affected plants.
The first serological methods used to detect and identify plant viruses were by reacting to the antibodies and the viruses (antigens) in liquid media and forming precipitates that could be viewed by the naked eye. Viral particles are polivalent, meaning that each viral particle can react with several molecules of the antibody that is divalent. This reaction forms a lattice structure that forms the visible precipitate during growth (Mattews 1970). This is called a “precipitation” reaction. The reaction of the agglutination of chloroplasts and other cell organelles is when a drop of infective sap of a plant with an unknown virus is made to react with a drop of specific antisera of known viruses, placed in a tray, and observed under a dissection microscope. The union of the antibodies with the viral particles formed a structure (lattice) that, in its weaving, precipitates, along with chloroplasts and other cell organelles (Kleczkowski 1965).
Another widely used method in the 1960’s and 1970’s was double diffusion in agar, a method carried out on trays or Petri dishes, with great advantages such as the following: (i) the mixtures of the molecules of the antigens and antibodies are physically separated due to their degree of diffusion in the gel, and (ii) several antigens or antibodies can be compared, placed in neighboring cells on the same tray or sheet (Ouchterlony 1962). The antigens and antibodies diffuse one against the other in the agar, and after a while, a precipitation zone is formed when they reach an acceptable concentration, which can be visible to the naked eye, or by staining with protein dyes and photographed (Figure 1). The diffusion of the viral antigen in the agar depends strongly on the size and shape of the virus. Isometric viruses diffuse in a satisfactory manner, although rod-shaped viruses diffuse slower or not at all. Splitting by sonification helps the diffusion in agar of the latter and the formation of the precipitate (Tomlinson and Walkey 1967). The main disadvantage of these models is the large volume of antiserum used in each detection reaction.

Figure 1 Antigens and antibodies confront each other when placing them in circular adjacent holes or wells made in the agarose so they form precipitation lines between them. Taken from: http://mesa54dinmuno.blogspot.mx/2009/05/tecnicas-inmunologicas.html.
The serological detection methods of precipitation in liquid media were used for approximately 20 years and progressively replaced by enzyme-linked immunosorbent assays, or ELISA. Clark and Adams (1977) showed that the ELISA method could be effectively applied to the detection of plant viruses. From that moment to date, this method has been widely used. Many variants of the basic procedure have been developed in an attempt to optimize the test for particular purposes.
The method is very cost-effective in the use of the reagents, and quickly adapted to qualitative and quantitative measurements. It can be applied to viruses of several types of morphologies and purified preparations or raw extracts (Figure 2). It is very sensitive, since it can detect concentrations of 1 - 10 ng/ml. The most common applicable methods of ELISA are: ELISA on plates; ELISA on membranes (immunobloting), using polyclonar or monoclonal antisera; Immunosorbent Electron Microscopy (ISEM) and immunochromatographic or Strip Tests (Hull, R 2014).

Figure 2 Jalapeño peppers affected by the synergy of the Cucumber mosaic virus (CMV) and the Tobacco etch virus (TEV), detected by the plate ELISA technique, in a screening for the 13 most common viruses using the macerate of the infected plant.
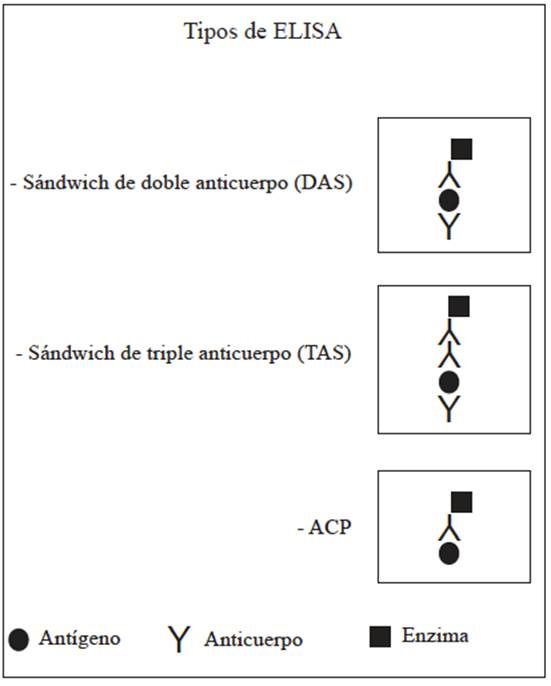
The most commonly used Plate ELISA types in plant pathology are: double antibody sandwich ELISA (DAS), Triple antibody sandwich ELISA (TAS), and antigen coated plate ELISA (ACP) (Figure 3).
The ELISA methods on membranes in its most common methods, immuno dot and inmuno printing, is printing on a nitrocellulose or nylon membrane of one microdrop (applied in microliters) of a plant extract or the print of a cut of a leaf, petiole, stem, or other plant tissue, directly on the membrane, respectively. In the ELISA methods on plates, the color is developed with a soluble substrate, and on the ELISA on membranes, it is replaced by a precipitant substrate that is insoluble and adheres to the membrane and it is read with the naked eye using a reflectance densitometer.
The direct printing of the tissue on the membrane (immuno printing) has several advantages: 1).- it provides detailed information of the distribution of the virus in the tissue; 2).- there is no need to macerate tissues, and therefore, the viruses restricted to certain tissues do not dilute, with the logical increase in sensitivity; 3).- the technique is perfectly applied to field samples and immuno-printing can be carried out on the field, without the need of taking the samples to the laboratory. In the Immunosorbent Electron Microscopy ELISA (ISEM), the sensitivity of the detection of the virus increases significantly under an electron microscope.
Immunochromatography, or reactive strip, is the quickest test currently used to detect countless plant pathogens, and it is produced by most, if not all, international diagnosis kit suppliers. There are ImmunoStrips to detect dozens of plant pathogens that are used directly by farmers in quick field tests, the results of which are obtained mere minutes after the tests have been performed (Figure 4). Serology is still being used to date through different types of ELISA, the most commonly used diagnosis methods for the detection of virosis in humans, animals, and plants.

Figure 4 Begonia affected by the Impatiens necrotic spot virus (INSV) detected using the ELISA field test - immunochromatography, in 5 minutes of exposure of the reactive strip to the infected sap.
Although serological methods are the most commonly used in viral diagnoses, they have certain disadvantages. They are based on the antigenic properties of the protein covers of the viruses, which accounts for only approximately 10% of the total genome of the virus (Gould and Symons 1983) and does not take the rest of the viral genome into account. The methods based on the detection of the nucleic acids have the advantage of being able to be directed to any region of the viral genome to be used in the diagnosis technique. Likewise, there are situations in which the immunological methods have a limited application, particularly for the detection of viroids, satellite RNAs, viruses that lack capsids (for example: Groundnut rosette virus (GRV) genus Umbravirus (http://viralzone.expasy.org/645), the NM-form of tobacco rattle virus), viruses with extremely diverse serotypes (for example, Indian and African Peanut clump virus and Tobacco rattle virus), and viruses that are poor immunogens, or that are dirrifult to purify.
Detection by molecular methods
The detection of viruses by molecular methods can be used when there is knowledge of at least a part of the virus genome sequence (Joo jin J 2014).
Hybridization of nucleic acids
This is used mainly with two purposes: to determine the relationship degree between two sequences of nucleic acids, for example, to viruses, and for the detection of viruses or other pathogens. The diagnoses of viruses by the hybridization of nucelic acids are carried out by immobilizing the test nucleic acid in membranes of nitrocellulose or nylon.
The dot or spot hybridization test is a commonly used technique in the detection of plant viruses (Owens and Dinner 1984). The process implies the solid-liquid hybridization, where: a).- the test nucelic acid (the viral nucelic acid of the sample) is placed and immobilized on a positively charged nitrocellulose or nylon membrane, b).- the union-free sites of the membrane are then blocked with a non-homologous DNA (generally salmon sperm or DNA from a calf thymus) or with protein (generally bovine albumn, egg albumin or milk casein), c).- allow the hybridization to take place between the union of the viral nucleic acid and the probe, which is free in the hybridization solution, d).- the excess probe which it did not hybridize is removed by a series of washings with a particular astringence, e).- the test sequence is detected by the reporting molecule used in the hybridized probe.
The radioactive probes that use 32P as a reporter are practically discontinued due to the short life and the danger of radioactive 32P, and have been replaced by non-radioactive probes, the most widely used of which, in virology, is the Dioxigenin (DIG)/antiDIG system. In this system, the membrane is exposed, post-hybridization, to antiDIG antibodies joined to the enzyme alkaline phosphatease or peroxidase. The signal is produced when adding the adequate substrate the results in a precipitated chromogenic product.
Polymerase chain reaction (PCR)
In 1986, Dr. Kary Mullis developed PCR, a molecular biology that amplifies a large number of copies of a particular DNA fragment. This technique is based on the natural property of DNA polymerases to replicate DNA, which requires four steps: a).- denaturalization at a high temperature (normally between 94 and 95°C) to separate the DNA strands; b).- annealing of the first two primers to their complementary sequence in their two DNA strands, the temperature of which depends on the size and the nucloetidic composition of the primer; c).- the extension of the primer to form de complementary chain by the polymerase DNA, normally at 72 °C, and d).- the final extension for 5 or 10 minutoes with the same temperature (Naidu and Huges, 2001). In each cycle, the new DNA strands act as a template for the new cycles, and therefore repeating the first three steps 30 or 40 times in an automatic thermocycler, the amount of new amplified DNA strands add up to millions and can be analyzed in an agarose gel, staining the DNA with ethidium bromide, which reveals the amplification performed; this technique is called end-point PCR. The speed, specificity, and the versatility of PCR made it more adequate and the most widely used in many areas of molecular biology and the most attractive in the diagnosis of viral diseases in plants (Rodriguez and Barrera 2004) (Figure 5).

Figure 5 Leaf of melon plant affected by Begomovirus detected by the end-point PCR technique, using primers specific for the genus Begomovirus.
Nested PCR is used to increase the sensitivity of the PCR. Here, two amplification rounds are used with different pairs of primers in each. First, an amplification is carried out with the external primers and another amplification is carried out with the product of that amplification, using internal primers, therefore increasing the sensitivity of the technique. This technique is used when the concentration of the virus or another pathogen is very low, and it is very commonly used to detect Phytoplasmas.
The multiplex PCR is another variation of the traditional end-point PCR, which can be used to detect 2 or more viruses or 2 or more different fragments of white DNA in the same reaction. The amplification is carried using 2 or more pairs of primers and increasing the reagents of the reaction proportionally, saving time in the detection of multiple pathogens.
If the viruses to be detected belong to an RNA genome, PCR has a previous step. The RNA strand must be retrotranscribed in complementary DNA with an enzyme called retrotranscripatase or reverse trascriptase. Once transcribed in the complementary DNA, a conventional PCR is carried out for its amplification, and it is known as RT-PCR.
The PCR technique has also evolved to avoid the use of agarose gel, like the end-point. The Real Time PCR or quantitative PCR (qPCR) combines the steps of DNA amplification in a single trial and avoids having to prepare electrophoresis gels to detect the amplified products. In 1966, Le Pecq and Paoletti reported that ethidium bromide (EtBr) intercalante of the double chain DNA fluoresces under UV light. This property was used to record the accumulation of DNA using a videocamera. This simple reaction, combined with the video taken, allowed the real-time birth of PCR, which combines the enormous sensitivity of the PCR technique with the precision that ensures the “in situ” monitoring of the products generated by this reaction in time. The qPCR technique can use general fluorophores or unspecific for the union of DNA, such as ethidium bromide or SYBR Green I, the latter of which is the most widely used reporter, a positively charged molecule that, when in solution, does not fluoresce. However, when it joins the minor groove of the DNA, its fluorescence increases 1000 times. The specific fluorescence methods start from different principles to the unspecific ones, and they have in common that the fluorescent signal is only emitted when detecting the amplified products (Temay et al., 2013). The specific methods follow the FRET (Fluorescense Resonance Energy transfer) principle, which consists of the transfer of energy between two fluorophores: a donor (reporter) and an acceptor (quencher), which emir fluorescences with different wavelengths. When the reporter and the quencher are near, the quencher absorbs all the fluorescence of the reporter. When this pair of molecules separates, the fluorescence of the reporter cannot be absorbed by the quencher, and could consequently be detected by the photodetector. (Rodríguez, M. and William, R. 2006.; Estefanía et al., 2015).
Digital PCR (dPCR), unlike real time PCR (qPCR) requires no reference standards ore calibration curves for the quantification of nucleic acids, but rather produces an accurate quantification of nucleic acids. In dPCR, the PCR sample is divided into thousands of nanodrops and after amplification, the drops that contain the sequence of the target DNA are detected by fluorescence as positive, and those without fluorescence, as negative. The Poisson statistical analysis of the number of nanodrops gives an exact count of the target DNA, and this helps count the charge of viruses or another pathogen or of any other white DNA in a sample (Biorad. Introduction to Digital PCR. Taken from http://www.bio-rad.com/es-mx/applications-technologies/introduction-digital-pcr). The disadvantage of the PCR techniques mentioned is that they all require a thermocycler and a well-equipped lab, as well as staff trained to carry out a diagnosis.
Isothermal DNA amplification is a methodology that is carried out at a constant temperature, and therefore, unlike conventional PCR does not require the use of a thermocycler or highly-equipped labs. Several molecular strategies have been used to achieve this amplification. The following is a list of their names and their acronyms: Loop-mediated isothemal amplification, LAMP; Strand-displacement amplification, SDA; Helicase-dependent amplification, HDA; Rolling-circle amplification, RCA; Nucleic acid sequence-based amplification, NASBA; and Recombinace polymerase amplification, RPA. Several companies from different countries are producing commercial diagnosis kits for the detection of phytopathogens with one or another of these technologies. Thus TwistDx, www.twistdx.co.uk, and Agdia, Inc. www.agdia.com use RPA technology; Ac Diagnostics, Inc. www.acdiainc.com uses NASBA technology; Diageneticx, Inc. www.bloomberg.com use LAMP technology, to mention a few. All these methodologies have advantages and disadvantages, according to the compilation made by Zaghloul, H and El-shahat, M (2014); the only one of these technologies that only has advantages is RPA.
Next-generation (high throughput) sequencing (NGS)
This technology was created in the past few years and was visualized as a groundbreaker in molecular biology in all its branches; its importance lies in that it has significantly reduced costs and time in comparison to Sanger traditional sequencing. The four main modern platforms that currently exist [Illumina Solexa] sequencing; Roche 454 sequencing; Ion torrent: Proton / PGM sequencing and SOLiD sequencing) produce large amounts of readings (between millions and billions) of short sequences, from 25 to 400 pb, although nowadays, there are platforms that read up to 700 pb.
As Figure 6 shows, the cost of sequencing a megabase between the years 2001 and 2013 was dramatically reduced. During this period, the first generation of sequencers was used from 2001 to 2007, and the second generation, from 2008 to 2013. The cost per sequenced megabase dropped from USD$8000 in 2001 to $700 for 2007, and later, to $0.10 in 2013 (Figure 6A). Similarly, the cost per sequenced DNA genome was USD $100, 000,000 in 2001 to $10, 000,000 in 2007 and to 8,000 in 2013 (Figure 6B).
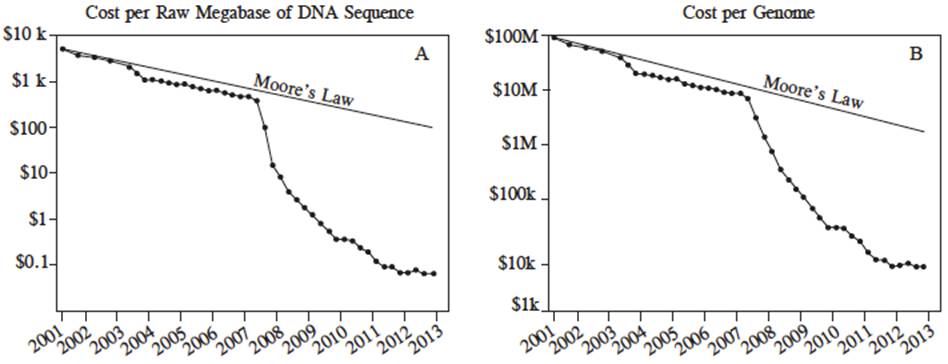
Figure 6 Cost in US dollars per megabase of DNA sequencing (A) and cost per genome (B) from July, 2001 to July 2013, estimated by the National Human Genome Research Institute, U.S. National Institues of Health, Bethesda, MD, USA. In this period, the first generation of sequencers was used (Sanger) from 2001 to 2007, after the NGS platforms were used between 2007 and 2013 (Marina et al., 2014).
This new technology came hand-in-hand with a new terminology, such as “Metagenomics,” defined as the application of new genomic technologies for the study of communities of microorganisms directly in their natural habitat, without having to isolate and cultivate them in the lab; or the “Sequencing of transcriptomes,” that includes the sequencing and analysis of all the complete mRNA’s and microRNA’s.
The NGS, combined with sophisticated bioinformatics, has changes the field of plant virology, mainly in the areas of genome sequencing, ecology, discoveries, epidemiology, transcriptomes, replication, detection, and identification. “Virome” refers to the viral load (viruses and viroids) that infect an organism, plant or animal, including vectors. Many new viruses and viroids have been identified using NGS technology, whether directly, sequencing their genomes, or even indirectly, assembling the small fragments of siRNA produced as a response to the infections caused by viruses and viroids (Marina et al., 2014).
LITERATURA CITADA
Brakke MK. 1951. Density gradient centrifugation: A new separation technique. Journal of the American Chemical Society. 73:1847-8. https://doi.org/10.1021/ja01148a508 [ Links ]
Brakke M K, 1953. Zonal separations by density-gradient centrifugation. Arch Biochem Biophys 45:275-90. Tomado de: Hull R, 2014. Matthew´s Plant Virology. Academic press, London. 778 p. https://doi.org/10.1016/S0003-9861(53)80005-6 [ Links ]
Barba M, Czosnek H and Hadidi A. 2014. Historical Perspective, Development and Applications of Next-Generation Sequencing in Plant Virology. Viruses. 2014 Jan; 6(1): 106-136. https://doi.org/10.3390/v6010106 [ Links ]
Clark MF and Adams AN. 1977. Characteristic of the microplate methods of enzyme-linked immunosorbent assay for the detection of plant viruses. Journal of General Virology. 34:475-483. https://doi.org/10.1099/0022-1317-34-3-475 [ Links ]
Estefanía C, Bayron J y Barreto A. 2015. PCR en tiempo Real. Biología Molecular-Camilo Herrera. Universidad de las Américas. Disponible en línea: https://es.slideshare.net/Alejatoxik/pcr-en-tiempo-real-55369307 [ Links ]
Gergerich RC and Dolja V. 2006. Introduction to plant viruses, the invisible foe. The Plant Health Instructor. https://doi.org/10.1094/PHI-I-2006-0414-01 [ Links ]
Gould AR and Symons RH. 1883. A molecular biological approach to relationship among viruses. Annual Review of Phytopathology. 21: 179-99. https://doi.org/10.1146/annurev.py.21.090183.001143 [ Links ]
Höltke HJ, Ankenbauer W, Mühlegger K, Rein R, Sagner G, Seibl R and Walter, T. 1995. The digoxigenin (DIG) system for non-radioactive labelling and detection of nucleic acids--an overview. Cell Mol Biol (Noisy-le-grand). Nov; 41(7):883-905 [ Links ]
Tomado de: Henson JM and French R. 1993. The polymerase chain reaction and plant disease diagnosis. Annual Review of Phytopathology 31: 81-109. https://doi.org/10.1146/annurev.py.31.090193.000501 [ Links ]
Hull R. 2014. Matthew´s Plant Virology. Elsevier, Academic Press. 1001 p. https://doi.org/10.1016/B978-0-12-384871-0.00023-6 [ Links ]
Joo-jin J, Ho-jong J and Jaejong N. 2014. A Review of Detection Methods for the Plant Viruses. Research in Plant Disease. 20:173-181. http://dx.doi.org/10.5423/RPD.2014.20.3.173 [ Links ]
Kelaniyangoda DB and Madhubashini LWM. 2008. Indicator plants: Tools for Detecting Papaya Ring Spot Potyvirus and Cucumber Mosaic Cucumovirus. Journal of Food and Agriculture. Vol.1(2) pp.64-69. http://doi.org/10.4038/jfa.v1i2.1800 [ Links ]
Kleczkowski A. 1965. A study of the effects of salt and pH on precipitation of antigen-anibody compounds, Immunology 8: 170-181 [ Links ]
Mattews 1970. Plant Virology. Academic press, London . 778 p. http://doi.org/10.4038/jfa.v1i2.1800 [ Links ]
Lister RM. 1966. Possible relationship of virus specific products of tobacco rattle virus infection. Virology. 28: 350-353. https://doi.org/10.1016/0042-6822(66)90161-9 [ Links ]
Lister RM. 1968. Functional relationship between virus-specific products of infection by viruses of the tobacco rattle type. Journal of General Virology. 2: 43-58. https://doi.org/10.1099/0022-1317-2-1-43 [ Links ]
Naidu RA and Hughes JdA. 2001. Methods for the detection of plant virus diseases. Plant Virology in Sub-Saharan Africa [ Links ]
Kumlachew A. 2015. Detection of Diseases, Identification and Diversity of Viruses: A Review. Journal of Biology, Agriculture and Healthcare. Vol.5, No.1. Disponible en línea: http://www.iiste.org/Journals/index.php/JBAH/article/view/19521. DOI: 10.1007/978-81-322-0813-6 [ Links ]
Outcherlony O. 1962. Diffusion-in-gel methods for immunological analysis II. Progr. Allergy. En: Mattews 1970. Plant Virology. Academic press, London. 778 p. https://doi.org/10.1159/000313795 [ Links ]
Owens RA and. Diener TO. 1984. Spot hybridization for the detection of viroids and viruses. Pages 173-189. In: Methods in Virology Vol. VII, edited by K. Maramorosch and H. Koprowski. Academic Press, New York, USA. https://doi.org/10.1016/B978-0-12-470207-3.50012-7 [ Links ]
Piepenburg O, Williams, CH, Stemple DL and Armes NA. 2006. DNA Detection Using Recombination Proteins. PLoS Biol 4(7): e204. https://doi.org/10.1371/journal.pbio.0040204 [ Links ]
Purdy HA. 1928. Immunologic reactions with tobacco mosaic virus, Proc. Soc. Exp. Biol. and IVied., XXV, 702-703. En: Scholthof, K-B. G. 2008. Tobacco Mosaic Virus: The Beginning of Plant Pathology. Online. APSnet Features. https://doi.org/10.1094/APSnetFeatures-2008-0408 [ Links ]
Rodríguez IPS y Barrera HAS. 2004. La reacción en cadena de la polimerasa a dos décadas de su invención. Ciencia UANL/Vol VII No.3: 323-35. Disponible en línea: http://eprints.uanl.mx/1584/1/art_cadena.pdf [ Links ]
Rodríguez M y William R. 2006. PCR en Tiempo Real. Métodos químicos-físicos en Biotecnología. IBT-UNAM. 55p. Disponible en línea: http://www.ibt.unam.mx/computo/pdfs/met/realtime_pcr.pdf [ Links ]
Roistacher CN. 1991. Graft-transmissible Diseases of Citrus. Handbook for detection and diagnosis. IOCV, Riverside and FAO, Rome. 286 pp. Disponible en línea: https://archive.org/details/bub_gb_9zY1uFGchAgC [ Links ]
Salazar Luis 1995. Los virus de la papa y su control. CIP. Perú. 226 p. disponible en línea: https://books.google.com.mx [ Links ]
Tammy DL, Ibarra C y Velasquillo C. 2013. Fundamentos de la reacción en cadena de la polimerasa (PCR) y de la PCR en tiempo real. Investigación en discapacidad. Vol. 2, Num 2: 70-78. Disponible en línea: http://www.medigraphic.com/pdfs/invdis/ir-2013/ir132d.pdf [ Links ]
Tomlinson JA and Walkey DGA. 1967. Effects of ultrasonic treatment on TurnipMosaic Virus and Potato Virus X. Virology. 32: 267-278. https://doi.org/10.1016/0042-6822(67)90276-0 [ Links ]
Welkin EJ (tomado de: Scott C. Weaver, Mark Denison, Marilyn Roossinck and Marco Vignuzzi). 2016. Virus Evolution: Current Research and Future Directions. Caister Academic Press. 366 p. https://doi.org/10.21775/9781910190234 [ Links ]
Zaghloul H and El-shahat M. (2014). Recombinase polymerase amplification as a promising tool in hepatitis C virus diagnosis. World Journal of Hepatology. 6(12): 916-922. https://doi.org/10.4254/wjh.v6.i12.916 [ Links ]
Received: June 06, 2017; Accepted: August 14, 2017
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
