











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
A diferencia de otros patógenos vegetales, el manejo de las enfermedades virales, basados en métodos directos, como el uso de viricidas para su control, no se han desarrollado a la fecha, por lo tanto, las enfermedades virales se combaten con estrategias indirectas, como el combate al insecto vector, eliminación las plantas enfermas, evitar la siembra de semillas infectadas. Por lo anterior, los métodos de detección e identificación de virus son críticos para el manejo de las enfermedades virales. Los métodos de detección deben de ser lo más convenientes, efectivos, específicos y rápidos (Joo-jin J. 2014) y son reconocidos como el instrumento básico de la virología, porque la exactitud de los estudios dependen directamente de la sensibilidad de éstas técnicas (Salazar 1995).
Los virus han co-evolucionado por millones de años hospedando a los tres dominios de las formas vivientes, arqueobacterias, Bacterias y Eucariotes (Gergerich y Dolja 2006), sin embargo su conocimiento es muy reciente, el estudio de los virus comienza con los trabajos de Mayer en 1886, quien trasmitió mecánicamente el Tobacco mosaic virus (TMV) de planta enferma a planta sana; Iwanowski en 1892, demostró que la savia de dichas plantas, seguía infectiva después de pasar por un filtro de porcelana a prueba de bacterias y Beijerinck en 1898 fue quien describió al agente causal como “Contagium vivum fluidum”, e introdujo por primera vez la palabra “virus”, para diferenciarlo del fluido contagioso corpuscular que contenía bacterias u esporas de hongos, dando así, inicio a la ciencia de la Virología (Hull R. 2014). Desde ese entonces, los investigadores han buscado como estudiar los virus para poderlos detectar en sus hospederos, identificar y clasificar todos los virus nuevos que se fueron descubriendo en ésta naciente rama de la ciencia.
Detección por Sintomatología
La sintomatología producida por los virus en las plantas, fue la primera forma de detectar e identificar los virus a finales del siglo XIX y su nombre fue asociado con los síntomas que producía, por ejemplo: Tobacco mosaic virus (TMV), Papaya ringspot virus (PRSV), etc., sin embargo muy pronto se dieron cuenta los investigadores, que muchas variantes del mismo virus producían síntomas muy diferentes y por otro lado, muchos virus diferentes producían síntomas muy similares y aunado a ello, las plantas también exhibían síntomas parecidos a virosis como respuesta a condiciones desfavorables del clima, balance nutricional de minerales del suelo, infecciones por patógenos no virales, daño causado por insectos, ácaros, nematodos. Aunque la sintomatología proveía una vital información sobre las enfermedades virales, se requería mucha experiencia de campo para tomar decisiones basados solo por la sintomatología para identificar un virus, generalmente es necesario que las inspecciones de campo sean acompañadas por otras pruebas para la correcta diagnosis de una infección viral (Naidú y Hughes. 2001).
Durante el período (1900 al 1935) la atención fue enfocada a la descripción e identificación por la sintomatología de la enfermedad, tanto macroscópicamente, como anormalidades ocasionadas dentro de las células vistas con microscopía de luz, llamadas inclusiones virales. Estos eran los únicos aspectos que se podían estudiar con las técnicas que tenían disponibles; todo era confusión, lo único que se sabía de los virus, era que éstos eran muy pequeños y causaban enfermedades. Holmes en 1929 demostró que las lesiones locales producidas por inoculación mecánica en ciertos huéspedes, podía ser usado para cuantificar cuantitativamente la cantidad de virus infectivos, esta técnica permitió estudiar algunas de las propiedades de los virus, como: Punto de inactivación térmica, punto de dilución máxima y longevidad in vitro y facilitó el camino para el futuro aislamiento y purificación de los virus.
Usando la técnica de trasmisión mecánica, muchas de las enfermedades de los vegetales ocasionados por virus filtrables fueron descritas de 1900 a 1935, sin embargo, pronto se encontró que muchas enfermedades con síntomas similares a enfermedades virales, no se trasmitían mecánicamente y el criterio de filtrabilidad no se podía aplicar, sin embargo su naturaleza infecciosa se podía comprobar por transmisión por injerto o por vectores, en esa forma, muchas de las enfermedades si fueron causadas por virus que no se trasmitían mecánicamente, pero muchas otras, con síntomas de amarillamientos o escobas de bruja, que fueron atribuidas a virus, después se comprobó, que eran causadas por Fitoplasmas o Spiroplasmas u otras bacterias no cultivables y no trasmisible mecánicamente.
Detección por Plantas diferenciales
En 1931 Smith trabajó con virus en papa y demostró que la sintomatología podía ser causada por la combinación de varios virus, en esta forma, utilizando plantas diferenciales y transmisión por insectos, pudo separar el Potato virus X (PVX) del Potato virus Y (PVY), al trasmitir éste último mediante el áfido Myzus persicae que no trasmitía el PVX y por inoculación mecánica en Datura stramonium separó el virus PVX, porque ésta planta es inmune al virus Y; en ésta forma pudo trabajar con virus puros, para determinar su rango de hospederos y por primera vez pudo observar las diferencias en virulencia de los aislamientos de un mismo virus por las diferencias en la sintomatología producida. Hasta ese momento, el uso de plantas diferenciales y su rango de hospederos, usando inoculación mecánica o por injerto o mediante el uso de vectores, era la única forma de identificar los virus en vegetales. En los Cuadros 1, 2 y 3 se observan los diferentes síntomas en las plantas diferenciales que eran utilizados para la identificación de Papaya ringspot virus (PRSV), Cucumber mosaic virus (CMV) (Kelaniyangoda and Madhubashini 2008), y 10 diferentes virus en papa (Salazar 1995), en esa forma se realizaban cuadros comparativos de síntomas para todos los virus que podían ser identificados, utilizando trasmisión mecánica, vectores o por injerto. La detección de virus mediante plantas diferenciales se sigue utilizando en muchos frutales como cítricos, pomáceos y rosáceos (Roistacher 1991).
Cuadro 1 Rango de hospederos y sintomatología para el diagnóstico del PRSV (tres semanas postinoculación).

x MC= Mancha clorótica; DF= Distorsión foliar; A= Achaparramiento; M= Mosaico; LL= Lesiones locales; ns= No síntomas
Cuadro 2 Rango de hospederos y sintomatología para diagnóstico del CMV

x DF= Distorsión foliar; MC= Manchas cloróticas; Mot= Moteado; M= Mosaico; LL= Lesiones locales; ns= No síntomas

Detección por Serología
El nacimiento de la serología para la identificación de los virus que afectan a los vegetales comenzó con el descubrimiento de Beale en 1928 demostrando que las plantas infectadas por el Tobacco mosaic virus (TMV) contenían un antígeno específico diferente a los de la planta sana, cuando ambas savias eran inyectadas en conejos diferentes, posteriormente Gratia en 1933, demostró que las plantas infectadas con diferentes virus, contenían antígenos específicos diferentes y por último Chester en 1935 y 1936 demostró que diferentes aislamientos de TMV y del PVX podían ser distinguidos serológicamente (Hull R 2004).
Conforme se fue avanzando en la metodología para la purificación de los virus, principalmente con el descubrimiento de Brakke (1951 y 1953) de la centrifugación en gradientes diferenciales usando diferentes concentraciones de cloruro de litio, cloruro o sulfato de cesio y posteriormente, con el desarrollo de la centrifugación en gradientes de sacarosa de 10% al 40%, sobre el cual se colocaba la savia infectiva (Lister, 1966 y 1968), el virus se separaba formando una banda donde su densidad igualara a la densidad de la sacarosa. Con los virus purificados se produjeron los antisueros específicos para cada uno de los virus purificados y que servían ya, para la identificación diferencial de los diferentes virus que afectaban a los vegetales.
Los primeros métodos serológicos utilizados para la detección e identificación de virus vegetales fueron reaccionando los antisueros y los virus (antígenos) en medios líquidos y formando precipitados que podían ser observados a simple vista. Las partículas virales son polivalentes, esto es, cada partícula viral puede reaccionar con varias moléculas del anticuerpo que es divalente, esta reacción forma una estructura látice que al crecer forma el precipitado visible (Mattews 1970), a ésta reacción se le llama “de precipitación”, la reacción de aglutinación de cloroplastos y de otros organelos celulares es cuando se hace reaccionar una gota de savia infectiva de una planta cuyo virus no se conoce con una gota de antisueros específicos de virus conocidos, colocadas en un portaobjeto y observada la reacción bajo microscopio de disección. La unión de los anticuerpos con las partículas virales formaba una trama (estructura latice) que en su entrelazado precipita junto a cloroplastos y otros organelos celulares (Kleczkowski 1965).
Otro método muy utilizado en la década de los 1960 y 1970 fue la doble difusión en agar, las grandes ventajas de éste método llevado a cabo en portaobjetos o cajas de Petri son: (i) las mezclas de las moléculas de los antígenos y anticuerpos están físicamente separados, por su diferente grado de difusión en el gel y (ii) se pueden comparar varios antígenos, o anticuerpos, colocados en celdas vecinas en una misma placa o cubreobjeto (Ouchterlony 1962). Los antígenos y los anticuerpos difunden uno en contra del otro en el agar y después de un tiempo se forma una zona de precipitación cuando alcanzan una concentración aceptable, la cual puede ser visible a simple vista o con tinción de proteínas y fotografiada (Figura 1). La difusión del antígeno viral en el agar depende fuertemente del tamaño y forma del virus, los isométricos difunden satisfactoriamente pero los de varilla difunde más lento o no difunden, el rompimiento por sonificación ayudan a la difusión en el agar de éstos últimos y la formación del precipitado (Tomlinson y Walkey 1967). El principal inconveniente de estos métodos, es el gran volumen de antisuero que se usa en cada reacción de detección.

Figura 1 Se enfrentan antígenos y anticuerpos colocándolos en hoyos o pozos circulares adyacentes hechos en la agarosa para que formen líneas de precipitación entre ellos. Tomado de: http://mesa54dinmuno.blogspot.mx/2009/05/tecnicas-inmunologicas.html.
Los métodos de detección serológica de precipitación en medio líquido fueron usados por aproximadamente 20 años y fueron progresivamente sustituidos por los métodos de ensayo de inmunoabsorción ligado a enzimas (enzyme-linked immunosorbent assay) o ELISA por sus siglas en inglés. Clark y Adams (1977) mostraron que el método de ELISA en placa podía ser efectivamente aplicado a la detección de virus vegetales, desde ése tiempo a la fecha, dicho método ha sido ampliamente usado. Muchas variantes del procedimiento básico han sido desarrolladas con el objetivo de optimizar la prueba para propósitos particulares.
El método es muy económico en el uso de los reactivos y rápidamente adaptado a las mediciones cualitativas y cuantitativas. Puede ser aplicado a virus de varios tipos de morfología y de preparaciones purificadas o de extractos crudos (Figura 2). Es muy sensible, pudiendo detectar concentraciones de 1 - 10 ng/ml. Los métodos aplicables de ELISA más comunes son: ELISA en placa; ELISA en membrana (immunobloting) empleando ya sea antisueros policlonales o monoclonales; inmunoadsorción-microscopía electrónica (ISEM por sus siglas en inglés) e inmunocromatografía o tiras reactivas (Immunochromatographic or Strip Tests) (Hull, R 2014).

Figura 2 Chile jalapeño afectado por la sinergia del Cucumber mosaic virus (CMV) y del Tobacco etch virus (TEV), detectados por la técnica de ELISA en placa, en un barrido (screening) para los 13 virus más comunes utilizando el macerado de la planta infectada.
Los tipos de ELISA en placa de uso más común en patología vegetal son: ELISA de sándwich de doble anticuerpo (DAS.- double antibody sándwich), ELISA de sándwich de triple anticuerpo (TAS.-Triple antibody sándwich) y ELISA de placa sensibilizada con antígeno (ACP.- antigen coated plate) (Figura 3).

Los métodos de ELISA en membrana en sus formas más comunes, inmuno-gota (immuno dot) e inmuno-impresion (immuno printing), es la impresión en una membrana de nitrocelulosa o nylon de una microgota (aplicada en microlitros) de un macerado vegetal o la impresión de un corte de una hoja, pecíolo, tallo u otro tejido vegetal, directamente sobre la membrana respectivamente. En los métodos de ELISA en placa, el desarrollo del color se realiza con un sustrato soluble y en la ELISA en membrana, se sustituye por un sustrato precipitante que es insoluble y que se adhiere a la membrana y su lectura se realiza a simple vista o con un densitómetro de reflectancia.
La impresión directa del tejido en la membrana (inmuno-impresión) tiene varias ventajas: 1).- da una información detallada de la distribución del virus en el tejido; 2).- no es requerido el macerado de tejidos y por ende, no se diluyen los virus restringidos a ciertos tejidos, con el aumento lógico de su sensibilidad; 3).- la técnica es perfectamente aplicada a muestras de campo y la inmuno-impresión puede realizarse directamente en el campo, sin necesidad de llevar la colecta de muestras al laboratorio. En la ELISA de inmuno-adsorción-microscopía electrónica (ISEM por sus siglas en inglés), aumenta significativamente la sensibilidad de la detección del virus bajo microscopio electrónico.
La inmunocromatografía o tira reactiva, es la prueba rápida más utilizada en la actualidad para la detección de innumerables patógenos vegetales y es producido por la mayoría si no es que todos de los proveedores internacionales de kits de diagnóstico. Existen inmunotiras para detectar decenas de patógenos vegetales que son utilizadas directamente por los agricultores en pruebas rápidas de campo, cuyos resultados se obtienen en minutos de realizada la prueba (Figura 4). La serología sigue siendo en la actualidad, atreves de los diversos tipos de ELISA, los métodos de diagnóstico más utilizados para la detección de virosis en humanos, animales y vegetales.

Figura 4 Begonia afectada por el Impatiens necrotic spot virus (INSV) detectado por la prueba de campo de ELISA-inmunocromatografía, en 5 minutos de exposición de la tira reactiva a la sabia infectada.
Aunque los métodos serológicos son los más utilizados en diagnosis de virus, tienen ciertas desventajas. Son basados en las propiedades antigénicas de la cubierta proteica de los virus, la cual representa aproximadamente solo el 10% del genoma total del virus (Gould and Symons 1983) y no toma en cuenta el resto del genoma viral. Los métodos basados en la detección de los ácidos nucleicos, tienen la ventaja de que pueden ser dirigidos a cualquier región del genoma viral para ser usado en la técnica de diagnóstico. Además, hay situaciones en que los métodos inmunológicos tienen aplicación limitada, en particular para la detección de viroides, ARNs satélites, virus que carecen de cápside (por ejemplo: Groundnut rosette virus (GRV) género Umbravirus (http://viralzone.expasy.org/645), the NM-form of tobacco rattle virus), virus que tienen serotipos extremadamente diversos (ejemplo: Indian y African Peanut clump virus y Tobacco rattle virus) y virus que son pobres inmunógenos o que son difíciles de purificar.
Detección mediante métodos moleculares
La detección de virus mediante métodos moleculares puede ser usada cuando existe conocimiento de cuando menos de parte de la secuencia del genoma del virus (Joo jin J 2014).
Hibridación de ácidos nucleicos
Se usa principalmente con dos propósitos, para determinar el grado de relación de dos secuencias de ácidos nucleicos, por ejemplo, de dos virus y para la detección de virus u otros patógenos. Las diagnosis de virus por hibridación de ácidos nucleicos, se lleva a cabo inmovilizando el ácido nucleico prueba, en membranas de nitrocelulosa o nylon.
El ensayo de hibridación por puntos (dot) o manchas (spot) es una técnica comúnmente utilizada en la detección de virus vegetales (Owens y Dinner 1984), el proceso implica la hibridación solido-líquido, donde: a).- el ácido nucleico prueba (el ácido nucleico viral de la muestra) es colocado e inmovilizado sobre una membrana de nitrocelulosa o de nylon positivamente cargada, b).- Los sitios libres de unión de la membrana son posteriormente bloqueados con un DNA no homólogo (generalmente esperma de salmón o DNA del timo de becerro) o con proteína (generalmente albumina bovina, albumina de huevo o caseína de leche), c).- permitir que la hibridación se lleve a cabo entre la unión del ácido nucleico viral y la sonda, la cual está libre en la solución de hibridación, d).- el exceso de sonda que no hibridó es removida mediante serie de lavados con determinada astringencia, e).- la secuencia prueba es detectada por la molécula reportera usada en la sonda hibridada.
Las sondas radioactivas usando como reportero al 32P están prácticamente en desuso por la vida tan corta y lo peligroso del 32P radioactivo y han sido reemplazadas por sondas no radioactivas y de ellas la que más se usa en virología es el sistema Dioxigenina (DIG)/antiDIG. En este sistema la membrana es expuesta post-hibridación a anticuerpos antiDIG unidos a la enzima fosfatasa alcalina o peroxidasa. La señal es producida al agregar el sustrato adecuado que resulta en un producto cromógeno precipitado.
Reacción en cadena de la polimerasa (PCR)
En 1986 el Dr. Kary Mullis desarrolló la PCR, técnica de Biología Molecular que amplifica un gran número de copias de un fragmento de ADN particular. Esta técnica se fundamenta en la propiedad natural de la ADN polimerasas para replicar hebras de ADN, para lo cual se emplean cuatro pasos: a).- desnaturalización a alta temperatura (normalmente entre 94 ó 95 °C) para separar las hebras del ADN; b).- anillado de los dos primers a su secuencia complementaria en sus dos hebras de ADN y cuya temperatura depende del tamaño y composición nuceotídica del primer; c).- la extensión del primer para formar la cadena complementaria por la ADN polimerasa, normalmente a 72 °C y d).- la extensión final durante 5 ó 10 minutos con la misma temperatura (Naidu y Huges, 2001). En cada ciclo las nuevas hebras de ADN sirven de plantilla para los nuevos ciclos por lo que repitiendo los primeros tres pasos 30 ó 40 veces en un termociclador automático, la cantidad de nuevas hebras de ADN amplificadas suman millones y pueden ser analizadas en un gel de agarosa tiñendo el ADN con bromuro de etidio que revela la amplificación realizada, a ésta técnica se le llama PCR de punto final. La velocidad, la especificidad, la versatilidad de la técnica de la PCR la hicieron la más adecuada y la más utilizada en muchas áreas de la Biología Molecular y la más atractiva en el diagnóstico de las enfermedades virales de los vegetales (Rodriguez y Barrera 2004) (Figura 5).

Figura 5 Hoja de planta de melón afectado por un Begomovirus detectado mediante la técnica de PCR punto final, usando primers específicos para el género Begomovirus.
Para aumentar la sensibilidad de la PCR, se usa la PCR Anidada, conocida también como Nested PCR, en ella se usan dos rondas de amplificación con distintos pares de primers en cada una, primero se realiza una amplificación con los primers externos y con el producto de ésa amplificación se realiza otra amplificación utilizando primers internos, aumentando así la sensibilidad de la técnica. Ésta técnica se usa cuando la concentración del virus u otro patógeno es muy baja y es muy usada para detectar Fitoplasmas.
La PCR multiplex es otra variante de la PCR tradicional (punto final), mediante la cual se pueden detectar 2 ó más virus o 2 ó más distintos fragmentos de ADN blanco en la misma reacción. La amplificación se realiza utilizando 2 ó más pares de primers y aumentando proporcionalmente los reactivos de la reacción, ahorrando tiempo en la detección múltiple de patógenos.
En caso de que los virus a detectar sean de genoma de ARN, la PCR tiene un paso previo, se tiene que retrotranscribir la hebra de ARN, en ADN complementario, mediante la utilización de una enzima llamada retrotranscripatasa o trascriptasa inversa. Una vez transcrito en ADN complementario, se realiza una PCR convencional para su amplificación y se conoce como RT-PCR.
La técnica de la PCR, ha evolucionado también para evitar el uso del gel de agarosa como la del punto final. La técnica del PCR Tiempo Real o PCR cuantitativa (qPCR por sus siglas en inglés) combina los pasos de amplificación de ADN y la detección en un único ensayo y evita tener que preparar geles de electroforesis para detectar los productos amplificados. Desde el año 1966, Le Pecq y Paoletti reportaron que el bromuro de etidio (EtBr) intercalante del ADN de doble cadena fluoresce bajo la luz UV. Esta propiedad fue aprovechada para grabar la acumulación de ADN utilizando una videocámara. Esta sencilla reacción combinada con la videografía permitió el nacimiento del PCR en tiempo real, el cual combina la enorme sensibilidad de la técnica de PCR con la precisión que asegura el monitoreo in situ de los productos generados por esta reacción a través del tiempo. La qPCR puede utilizar fluoróforos generales o no específicos de unión al ADN, como el Bromuro de Etidio o el SYBR Green I, el reportero más usado es el SYBR Green, la cual es una molécula cargada positivamente que cuando está en solución, no emite fluorescencia, sin embargo cuando se une al surco menor del ADN su fluorescencia aumenta 1000 veces, o métodos de fluorescencia específicos parten de principios diferentes de los no específicos y tienen en común que la señal fluorescente es emitida solo al detectar los productos amplificados (Temay et al., 2013). Los métodos específicos siguen el principio FRET (Fluorescense Resonance Energy Transfer, por sus siglas en inglés), que consiste en la transferencia de energía entre dos fouoróforos: un donador (reportero) y un aceptor (apagador o quencher), los cuales emiten fluorescencia de diferente longitud de onda. Cuando el reportero y el apagador se encuentran próximos, el apagador absorbe toda la fluorescencia del reportero. Cuando éste par de moléculas se separa la fluorescencia del reportero no puede ser absorbida por el apagador y en consecuencia puede ser detectada por el fotodetector. (Rodríguez, M. y William, R. 2006.; Estefanía et al., 2015).
La PCR digital (dPCR), a diferencia de la PCR tiempo real (qPCR) no necesita estándares de referencia o curvas de calibración para la cuantificación de ácidos nucleicos, sino directamente produce una cuantificación precisa de ácidos nucleicos. En la dPCR, la muestra de PCR es dividida en miles de nanogotas y después de la amplificación, las gotas que contienen la secuencia del ADN blanco, se detectan por fluorescencia como positivas y las que no fluorescen como negativas. El análisis estadístico de Poisson del número de nanogotas positivas nos da una cuantificación exacta del ADN blanco, esto permite cuantificar la carga viral u otro patógeno o de otro ADN blanco cualquiera de una muestra (Biorad. Introduction to Digital PCR. Tomado de: http://www.bio-rad.com/es-mx/applications-technologies/introduction-digital-pcr). La desventaja de las técnicas de la PCR mencionadas, es que todas requieren de un termociclador y un laboratorio bien equipado y de personal especializado para realizar los diagnóstico.
La Amplificación de ADN Isotérmica es una metodología que se lleva a cabo a temperatura constante por lo que a diferencia de la PCR convencional no necesita del uso de un termociclador, ni de laboratorios muy equipados. Se han usado varias estrategias moleculares para lograr dicha amplificación , las enumero por su nombre en español, el originales en inglés con su acrónimo: Amplificación isotérmica mediada por bucle (Loop-mediated isothemal amplification, LAMP), Amplificación por desplazamiento de hebras (strand-displacement amplification, SDA), Amplificación dependiente de la Helicasa (Helicase-dependent amplification, HDA), Amplificación del circulo rodante (Rolling-circle amplification, RCA), Amplificación de secuencias de ácido nucleico (Nucleic acid sequence-based amplification, NASBA) y Amplificación de polimerasa recombinante (Recombinace polymerase amplification, RPA). Varias empresas de diferentes países están produciendo kits de diagnóstico comerciales para detección de fitopatógenos con una u otra de éstas tecnologías, así, TwistDx, www.twistdx.co.uk y Agdia, Inc. www.agdia.com usa la tecnología RPA; Ac Diagnostics, Inc. www.acdiainc.com usa la tecnología NASBA; Diageneticx, Inc. www.bloomberg.com usa la tecnología LAMP, por mencionar algunas. Todas éstas metodologías tienen ventajas y desventajas, según la recopilación realizada por Zaghloul, H y El-shahat, M (2014), la única de éstas técnicas que solo tiene ventajas es la RPA.
Next-generation (high throughput) sequencing (NGS) Segunda generación de secuenciadores
Es una tecnología que se ha desarrollado en los últimos años y está visualizada para ser un parte-aguas en la Biología Molecular en todas sus ramas, su importancia estriba en que ha bajado significativamente el costo y el tiempo con respecto a la secuenciación tradicional Sanger. Las cuatro principales plataformas modernas que hay de NGS; Illumina (Solexa) sequencing; Roche 454 sequencing; Ion torrent: Proton / PGM sequencing y SOLiD sequencing, producen grandes cantidades de lecturas (millones a miles de millones) de secuencias cortas de 25 a 400 pb, aunque recientemente ya hay plataformas que leen hasta 700 pb.
Como se puede observar en la Figura 6, el costo por secuenciar una megabase del año 2001 al 2013 fue reducido dramáticamente. Durante éste período fue usado la primera generación de secuenciadores del 2001 al 2007 y la segunda generación de secuenciadores del 2008 al 2013. El costo por megabase secuenciada fue reducido de $8000 (dólares americanos) en el 2001 a $700 para el 2007 y después a $0.10 en el 2013 (Figura 6A). Similarmente, el costo por genoma de ADN secuenciado fue de $100, 000,000 (dólares americanos) en el 2001 a $10, 000,000 en el 2007 y a 8,000 en el 2013 (Figura 6B).
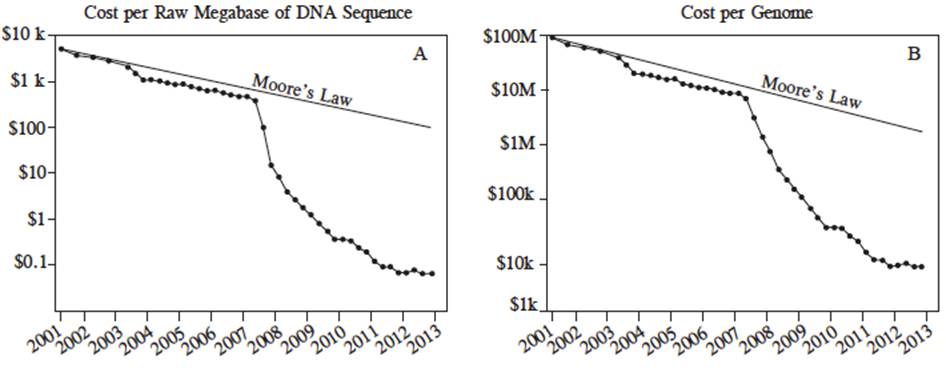
Figura 6 Costo en dólares americanos por megabase de ADN secuenciado (A) y costo por genoma (B) de julio de 2001 a julio del 2013 estimado por el National Human Genome Research Institute, U.S. National Institues of Health, Bethesda, MD, USA. Durante este período la primera generación de secuenciador fue usado (Sanger) del 2001 al 2007, después las plataformas de NGS fueron usadas del 2007 al 2013 (Marina et al., 2014).
Esta nueva tecnología ha traído consigo también una nueva terminología, como la “Metagenómica” definida como la aplicación de las nuevas tecnologías genómicas para el estudio de comunidades de microorganismos directamente en su ambiente natural, sin tener que ser aislados y cultivados en el laboratorio. “Secuenciado de transcriptomas” que incluye la secuenciación y análisis de todos los mARN completos y los microARN.
La NGS combinada con una bioinformática sofisticada ha cambiado el campo de la virología vegetal principalmente en las áreas de secuenciación de genomas, ecología, descubrimiento, epidemiología, transcriptomas, replicación, detección e identificación. “Viroma” se refiere a describir la carga viral (virus y viroides) que infectan a un organismo, vegetal o animal incluyendo a vectores. Muchos nuevos virus y viroides han sido identificados usando la tecnología de NGS ya sea en forma directa secuenciando sus genomas o incluso en forma indirecta, ensamblando los pequeños fragmentos de ARNsi que se producen como respuesta a las infecciones causadas por los virus y los viroides (Marina et al., 2014).

