











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
A variety of methods for the detection and identification of plant and fungal viruses are available to researchers and diagnosticians. For many years, serological methods and biological assays were commonly used in combination with viral nucleic acid detection. Today, nucleic acid-base approaches to virus detection such as polymerase chain reaction (PCR), together with sequencing, are widely used because of the high sensitivity and adaptation to the development of high throughput methods (Bonham et al., 2014). Recently, new technologies for virus detection and identification such as next-generation sequencing (NGS) have become available. NGS is a method with great potential for the detection and identification of plant viruses (Al Rwahnih et al., 2015; Bonham et al., 2014; Ho and Tzanetakis, 2013). For any nucleic acidbased method for RNA virus detection, efficient and practical RNA extraction protocols are needed. Many methods, including those of commercial kits, are currently available to extract single stranded RNA (ssRNA) from a wide range of biological tissues. Nevertheless, in some cases, the instability of ssRNA can be a factor affecting successful virus detection. The extraction, purification, and gel electrophoresis analysis of large (0.5-20.0 kb) viral double-stranded RNAs (dsRNAs) provide a complementary tool for scientists aiming to detect and identify plant viruses (Dodds et al., 1984; 1988; Morris and Dodds, 1979; Valverde et al., 1990b). Moreover, it also provides a source of RNA that can be used in other nucleic acid-based detection and identification methods for RNA viruses.
Large dsRNA has been recognized as genetic material in many plant, animal, fungal, and bacterial viruses (Libonati et al., 1980). Some biological functions of dsRNA include the capacity to induce interferon and the inhibition of protein synthesis in eukaryotes (Libonati et al., 1980). DsRNA is resistant to RNases under high ionic strength conditions. Under standard assay conditions (0.15 M NaCl, 0.015 M sodium citrate, pH 7), dsRNA is resistant to RNase A but not to RNases of the pancreatic type (Libonati and Sorrentino, 1992). Another unique property of dsRNA is that it binds to cellulose in ethanol-containing buffers. Optimally, dsRNA binds to fibrous cellulose at ethanol concentrations of about 16 % (Franklin, 1966; Morris and Dodds, 1979). The relatively high stability of dsRNA and the availability of extraction methods make the use of dsRNA practical for the detection and identification of RNA viruses.
In most plants and fungi infected with RNA viruses, dsRNAs can be found as part of: 1) genomic segments of dsRNA viruses; 2) replicative forms and replicative intermediates of single-stranded RNA viruses (ssRNA) (Buck, 1999; Horiuchi and Fukuhara, 2004; Jackson et al., 1971; Libonati et al., 1980; Nuss and Koltin, 1990; Zelcer et al., 1981); 3) subgenomic viral RNAs (sgRNAs) (Condit and Fraenkel-Conrat, 1979; Osman and Buck 1990); 4) satellite viruses (Klein and Reichman, 1970; Hillman et al., 2000; Valverde and Dodds, 1986); 5) satellite RNAs (Demler and de Zoeten, 1989; Liu et al., 2015); and 6) defective interfering RNAs (Burgyan et al., 1989; Hillman et al., 2000). Serological detection of dsRNAs from virus-infected plants and fungi has also been reported. An antiserum against polyinosinicpolycytidylic acid (polyi:c), a synthetic analog of double-stranded RNA, has been used to detect dsRNA from plants infected with Cucumber mosaic virus (CMV) and Citrus tristeza virus (CTV) and Penicillium chrysogenum infected with Penicillium chrysogenum virus using serological techniques (Aramburu et al., 1991). These techniques showed similar sensitivity for the detection of dsRNA as separation by polyacrylamide gel electrophoresis and silver staining. DsRNAs from Cowpea mosaic virus and Sugar cane mosaic virus have been detected in infected tissue extracts using serologically specific electron microscopy (Derrick et al., 1994). Monoclonal antibodies to dsRNA have been developed and used to detect Ground nut rosette virus in peanut (Schonborn et al., 1991). However, techniques based on serological methods require the development of the specific antibodies, a procedure that is more costly and lengthy than the direct visualization of dsRNA using electrophoresis or RT-PCR detection.
RNA viruses of plants and fungi
Viruses with ssRNA genomes make up more than 90 % of all known plant viruses. During replication in host cells, ssRNA viruses generate double-stranded replicative forms (RF) which consist of fully base-paired RNA molecules of the genomic RNA and replicative intermediates (Buck, 1999). They have been shown to occur in Tobacco mosaic virus (TMV) and Tobacco ringspot virus infected plants (Buck 1999; Derrick, 1978; German and de Zoeten, 1975; Jackson et al., 1971; Zelcer et al., 1981) and Oryza sativa endornavirus (Horiuchi and Fukuhara, 2004) among others. Many ssRNA viruses generate subgenomic RNAs (sgRNAs) to express internal open reading frames. These sgRNAs are synthesized either from prematurely terminated negative strands or, more commonly, by internal initiation on the negative-strand templates at specific promoter sequences (Miller and Koev, 2000). Subgenomic dsRNAs have been detected in plants infected with most RNA viruses and fungi infected with RNA mycoviruses (Condit and Fraenkel-Conrat, 1979; Kwon et al., 2007; Osman and Buck, 1990). Plant viruses with dsRNA genomes include members of the families Reoviridae, Partitiviridae, Amalgaviridae, Totiviridae, and Chrysoviridae, and their genomic dsRNA are readily detected from plant tissues (Antoniw et al., 1986; Chen et al., 2016; Karan et al., 1994; Li et al., 2013; Quito et al., 2011; Sabanadzovic et al., 2009; 2011; Wei et al., 2003). With the exception of theReoviridae, the other four families contain plant viruses that do not cause visible symptoms and have been named persistent viruses. In contrast, acute viruses cause symptoms and economically important plant diseases (Roossinck, 2010).
All major taxa of fungi have been shown to contain viruses. Although most fungal viruses described to date have dsRNA genomes, there are some with ssRNA genomes and one report of a ssDNA genome (Ghabrial et al., 2015). There are many citations in the literature of the presence of viral dsRNAs in fungi (Azzam and Gonsalves, 1999; Castillo et al., 2011; Day et al., 1977; Delye and Corio-Costet, 1998; Dickinson and Pryor, 1989; Herrero et al., 2009; Khalifa and Pearson, 2014; Kim et al., 2005; Kondo et al., 2016; Li et al., 2014; Shamoun et al., 2008); however, in most cases these viruses do not appear to affect the host’s phenotype (Enebak et al., 1994; Reyes et al., 2003). Only in some cases they have been associated with phenotypic and genotypic variations (Castanho et al., 1978; Ghabrial et al., 2015; Wei et al., 2003). The ability of some mycoviruses to induce hypovirulence has been used for biological control of their fungal hosts that are pathogenic to plants (Nuss and Koltin, 1990). The best known examples of mycovirus-induced hypovirulence are virus strains of the chestnut blight fungus Cryphonectria parasitica (Choi and Nuss, 1992; Day et al., 1977; Lee et al., 2006). Methods for the extraction and purification of dsRNA have been essential for the discovery, detection, and characterization of mycoviruses (Nuss and Koltin, 1990).
Satellite viruses, satellite RNAs, defective interfering RNAs, and viroids
Satellites are subviral agents that depend on a helper virus for their replication. Satellite viruses code for their own coat protein and their RNA is encapsidated. Satellite RNAs do not encode capsid proteins, but are packaged in a protein encoded by the helper virus. The size of most satellites associated with plant and fungal viruses range from 2.0-0.4 kb (Hillman et al., 2000; Kaper and Diaz-Ruiz, 1977; Demler and de Zoeten, 1989; Valverde and Dodds, 1987). Defective interfering RNAs (DIs) are RNAs that result from errors in virus replication (Pathak and Nagy, 2009). Satellite RNA viruses, satellite RNAs, and DI RNAs can attenuate or enhance the symptoms caused by their parent virus (Pathak and Nagy, 2009; Habili and Kaper, 1981). Some satellite viruses and RNAs associated with plant viruses accumulate relatively large quantities of dsRNA (Diaz-Ruiz and Kaper, 1977; Dodds et al., 1984; Habili and Kaper, 1981; Valverde and Dodds, 1986; 1987). In fungi, dsRNA satellites have been associated with several dsRNA viruses (Liu et al., 2015; Romanos et al., 1981), and satellite dsRNAs have been reported in Sclerotinia sclerotiorum (Liu et al., 2015). A satellite RNA of Ophiostoma novo-ulmi Mitovirus 3a in hypovirulent Isolates of Sclerotinia homoeocarpa (Deng and Boland, 2004), and a satellite and DIRNAs of Cryphonectria hypovirus 3-Grand Haven 2 (Hillman et al., 2000) have also been identified.
Viroids are some of the smallest known infectious agents. They consist of non-coding circular ssRNA molecules forming highly basepaired rod-like structures and are capable of causing economically important diseases in many crops worldwide (Kovalskaya and Hammond, 2014). Viroid derived dsRNAs have been described (Carbonell, et al., 2008).
DsRNA extraction and purification
Over the past 50 years, several methods for large dsRNA extraction from virus-infected plant, animal and fungal tissues, as well as bacteria have been reported in the literature (Akin et al., 1998; Balijja et al., 2008; Castillo et al., 2011; Delye and Corio-Costet, 1998; DePaulo and Powell, 1995; Diaz-Ruiz and Kaper, 1978; Franklin, 1966; Morris and Dodds, 1979, Morris et al., 1983; Okada et al., 2015; Tzanetakis and Martin, 2008; Valverde et al., 1990) (Table 1). Most of these methods are based on cellulose chromatography, some on enzymatic degradation or lithium chloride precipitation. Recently, Atsumi et al., (2015) developed a novel dsRNA isolation method using a recombinant dsRNA-binding protein.
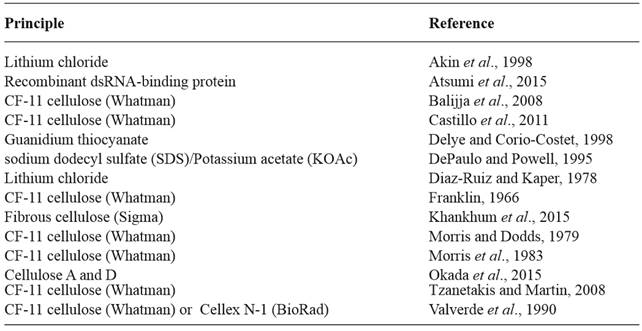
In 1979, Morris and Dodds developed a procedure for the isolation and analysis of dsRNA which has been widely used for the detection, identification, and characterization of plant viruses. It consists on the selective adsorption of dsRNA to fibrous cellulose at low ethanol concentrations and elution with ethanol-free buffer. A schematic diagram summarizing the steps involved in the method is shown in Figure 1. Although developed over 40 years ago, it is still in use today although it has been modified and improved (Khankhum et al., 2015; Valverde et al., 1990).
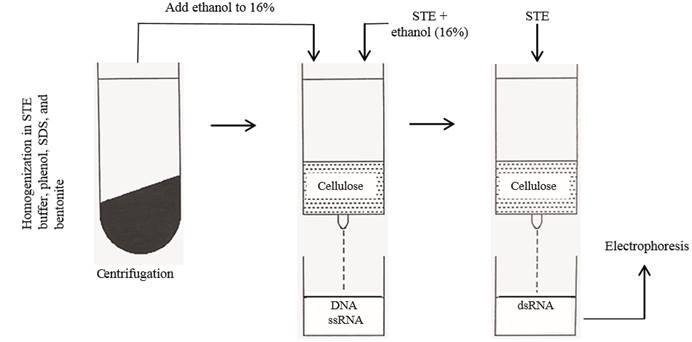
Figure 1. Schematic diagram of the steps involved in dsRNA extraction and purification. (Modified from Valverde et al., 1990). STE: Sodium, Tris and EDTA buffer (100 mM NaCl, 10 mM Tris-Cl, pH 8.0 and 1 mM EDTA); SDS: sodium dodecyl sulfate.
Homogenizing by grinding fresh, infected tissue, often with mortar and pestle using liquid nitrogen, is the first step in the extraction of dsRNA. However, buffered sap extracts, fresh or lyophilized fungal mycelium, dried spores, and desiccated plant tissue (including those infected with biotrophic fungi) have also been used with successful results (Morris et al., 1983; Khankhum et al., 2015; Kousik et al., 1994; Valverde et al, 1990). Other practical and efficient approaches for disrupting the tissue include bead beaters, tissue homogenizers, etc. Desiccated tissue has broad use because this method is conventionally used for the long-term storage of virus-infected tissue. It is also the way samples are exchanged between laboratories. Furthermore, biotrophic fungi infecting plant tissues are often stored in fungaria as pressed and dried specimens from which samples can be used to test for the presence of fungal viruses (Khankhum et al., 2015). In the case of plants, most extractions are done from foliar tissue. However, bark, stem, or vein tissue are recommended for phloem-limited viruses. For fungi, mycelium or spores can be used. Negative controls are highly recommended in any dsRNA extraction. The second step involves total nucleic acid extraction using buffer-saturated phenol. With some plant tissues and particular viruses buffer may be used instead of phenol. Nevertheless, phenol extracts usually yield cleaner samples. The third step involves binding of the dsRNA to fibrous cellulose. The cellulose used by Morris and Dodds (1979) and Franklin (1966) in their original papers was Whatman CF-11 (Whatman Inc., Clifton, NJ, USA). Because this cellulose is no longer commercially available, other cellulose types such as Cellex-N1 (Bio-Rad, Hercules, CA, USA) have been used in dsRNA purifications with some success (Okada et al., 2015; Valverde et al., 1990). Khankum et al., (2015) used fibrous cellulose (Sigma-Aldrich, St. Louis, MO, USA) to extract dsRNAs from virus-infected plants and fungi. This cellulose is readily available worldwide and a good alternative to CF-11. After ethanol precipitation, dsRNA samples must be treated with DNase to eliminate potential host DNA contamination. Host ribosomal RNA may interfere with the electrophoretic detection of dsRNAs ranging from 1.0-0.5 kb. Therefore, when the target dsRNAs are within this range, digestion with S1 nuclease is recommended. After that, analysis of dsRNAs can be conducted in 6 % polyacrylamide gels, which provide high resolving power for subgenomic dsRNAs or, more simply, using 1.21.5 % agarose gels. Traditionally, ethidium bromide has been the preferred stain, although silver nitrate staining has been used as well. More recently, staining with Gel RedTM (Huang et al., 2010) has become more common. GelRed does not have the health hazards associated with ethidium bromide and has higher sensitivity. Detailed information of the methodology developed by Morris and Dodds (1979) and data showing some of its applications has been published elsewhere (Dodds et al., 1984; 1988; Valverde et al., 1986; 1990b).
Initially, the focus of the dsRNA extraction protocols was on the electrophoretic analysis and evaluation of banding patterns for virus detection and, in some cases, identification. Although analysis of viral dsRNAs by gel electrophoresis is still widely used, currently most applications are in RT-PCR, molecular cloning, sequencing, genome assembly, and virus identification.
Virus detection and identification
In many cases, particularly with ssRNA viruses infecting plants that contain high amounts of phenolic compounds and carbohydrates, diagnosticians and researchers find it difficult to obtain pure preparations of ssRNA or virus to use in identification and characterization. An alternative is to purify dsRNAs that can be used for probe preparation, RT-PCR, molecular cloning, and sequencing.
In general, different families of RNA viruses infecting plants or fungi yield characteristic electrophoretic dsRNA banding patterns that differentiate them (Dodds et al., 1984; Valverde et al., 1986). DsRNA electrophoretic profiles have been reported for members of the families Closteroviridae (Dodds and Bar-Joseph, 1983), Potyviridae (Valverde et al., 1986), Bromoviridae (Valverde and Glascock, 1991), Endornaviridae (Fukuhara et al., 2006) and the genera Comovirus (Valverde et al., 1995), Tobamovirus, Potexvirus, and Carlavirus (Valverde et al., 1986). As mentioned earlier, dsRNA profiles generally consist of one (in the case of monopartite viruses), two (bipartite) or more (multipartite) full-length genomic bands (or RFs) and several other bands corresponding to subgenomic replicative forms of RNA viruses (Condit and Fraenkel-Conrat, 1979; Osman and Buck, 1990). Figures 2 and 3 show dsRNA profiles of different plant viruses resolved in polyacrylamide and agarose gels. In the case of fungal viruses, dsRNA electrophoretic profiles have not been evaluated as extensively as a tool to differentiate between families or genera. Nevertheless, as in the case of plant RNA viruses, electrophoretic profiles vary depending upon the virus species and mixed infections are common (Figure 4).
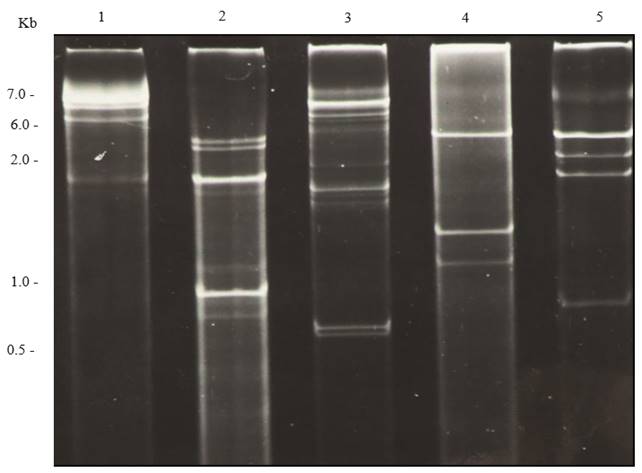
Figure 2. Polyacrylamide (6 %) gel electrophoresis of dsRNA extracted from plants infected with different viruses. Lane 1, Potato virus X; lane 2, Cucumber mosaic virus; lane 3, Tobacco mild green mosaic virus; lane 4, Tobacco necrosis virus; and lane 5, Alfalfa mosaic virus.
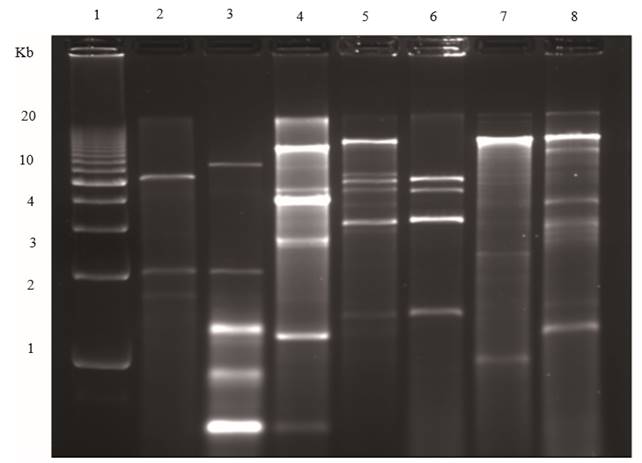
Figure 3. Agarose (1.2 %) gel electrophoresis of dsRNAs extracted from plants infected with different viruses. Lane 1, Molecular weight DNA ladder; lane 2, Tobacco necrosis virus; lane 3, Panicum mosaic virus and satellite virus; lane 4, Japanese holly fern mottle virus; lane 5, Tobacco mosaic virus, Alfalfa mosaic virus and Cucumber mosaic virus mixed infection; lane 6, Cucumber mosaic virus; lane 7,TMV; lane 8, Tomato mild green mosaic virus.
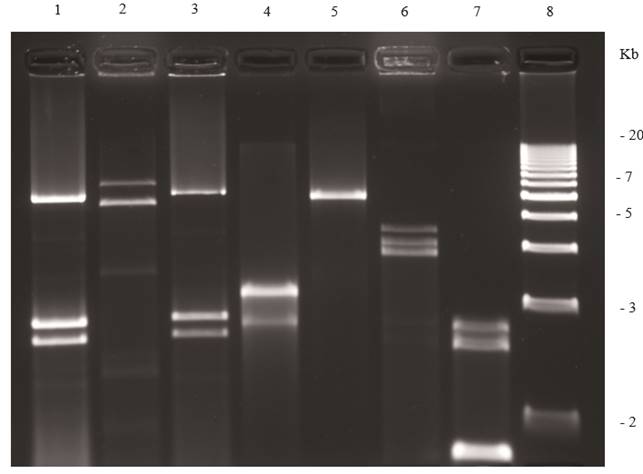
Figure 4. Agarose (1.2 %) gel electrophoresis of dsRNAs extracted from fungi. Lane 1, Penicillium digitatum (orange); lane 2, Corn smut; lane 3, Penicillium digitatum (mandarin); lane 4, Diaporthe phaseolorum isolate B; lane 5, an unidentified totivirus from D. phaseolorum isolate C; lane 6, Penicillium chrysogenum virus; lane 7, an unidentified virus from Penicillium stoloniferum; and lane 8, 1 kb molecular weight DNA ladder.
Extraction and electrophoretic analyses of dsRNA has been used widely to study important viruses such as Citrus tristeza virus (CTV) (Dodds and Bar-Joseph, 1983; Dodds et al., 1993) and viruses of C. parasitica, the causal agent of chestnut blight (Casthano et al., 1978; Day et al., 1977; Hansen et al., 1985; Lee et al., 2006). In the literature, there are numerous examples of the use of viral dsRNA to detect, identify, and study viruses, satellites, and DIs infecting plants (Aboughanem-Sabanadzovic et al., 2016; De la Torre, et al., 2014; 2016; Demler and Zoeten, 1989; Dodds, 1982; Falk et al., 1979; Okada et al., 2011; Valverde and Dodds, 1986; Valverde et al., 1986; Valverde et al., 1995; Ward et al, 2009) and fungi (Azzam and Gonsalves, 1999; Deng and Boland, 2004; Kousik et al., 1994; Feldman et al 2012; Pecina et al., 2000; Shamoun et al, 2008).
In plants, mixed viral infections often go undetected and can result in inadequate disease diagnoses. An advantage of dsRNAs analysis is that, unlike other virus diagnostic techniques, is nonspecific and therefore it is possible to visualize mixed viral infections (Figure 5). In fungi, mixed infections of mycoviruses are commonly detected by electrophoretic analysis of their dsRNAs (Chu et al., 2004; Herrero et al., 2012; Kousik et al., 1994). Another advantage of this technique is that in some cases, strains of the same virus can be differentiated by the mobility or the number of dsRNA fragments after gel electrophoresis. This has been shown with various strains of Tobacco mosaic virus satellite (Valverde and Dodds, 1987). Moreover, gel electrophoretic profiles of viral dsRNA have been used to differentiate strains of Tomato mosaic virus (Valverde et al., 1986), CMV (Valverde et al., 1990a), and CTV (Dodds et al., 1987). Nevertheless, in plants, members of the viral families Potyviridae and Luteoviridae generate low amounts of RF and subgenomic dsRNAs (Creamer and Falk, 1989; Dale et al., 1986; Valverde et al., 1986) making their detection by electrophoretic analysis impractical.
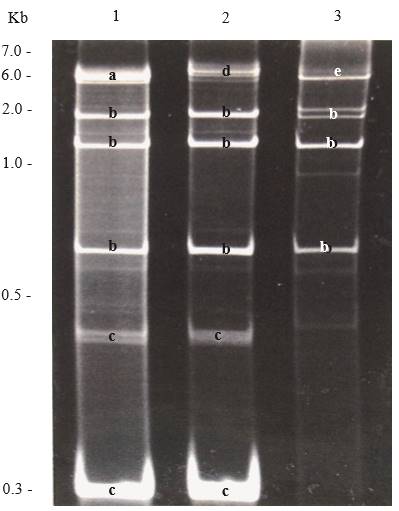
Figure 5. Polyacrylamide (6 %) gel electrophoresis showing dsRNAs extracted from plants with mixed viral infections. Lane 1, Potato virus X (PVX), Cucumber mosaic virus (CMV) and satellite RNA of CMV; lane 2, Dandelion latent virus (DLV), CMV and satellite RNA of CMV; and lane 3, Tobacco mosaic virus (TMV and CMV. Bands are labelled with letters according to the virus they belong; a, PVX; b, CMV, c, Satellite RNA of CMV; d, DLV, and e, TMV.
Molecular probes
Radioactive and non-radioactive probes have been produced by using dsRNA to generate and label ssRNA or cDNAs (Enebak et al., 1994; Jelkmann et al., 1989; Jordan and Dodds, 1983; Rosner et al., 1983; Valverde and Dodds, 1986; Valverde et al., 1994; Valverde et al., 1990b). Jordan and Dodds used TMV dsRNA to develop P32 endlabelled ssRNA probes and used them successfully in molecular hybridization experiments. Nonradioactive probes have also been prepared by direct labelling ssRNA using dsRNA as the starting material (Valverde et al., 1994). These non-radioactive probes were used to detect Oryza sativa endornavirus, Physalis mottle virus, Pepper cryptic virus 1, and an unidentified mycovirus from the soybean stem canker pathogen (Diaporthe phaseolorum). DsRNA extracts blotted onto membranes have been used to detect Sweet potato chlorotic stunt virus (SPCSV) in (Pio-Ribeiro et al., 1996), Fiji virus (Karan et al., 1994), Satellite tobacco mosaic virus (Valverde and Dodds, 1986), and a 12 kb hypovirus from C. parasitica (Enebak et al., 1994) in northern hybridization assays.
Reverse transcription PCR, cloning, and sequencing
Viral dsRNAs have been used to clone and sequence plant and fungal RNA viruses (Aboughanem-Sabanadzovic et al., 2016; Antoniw et al., 1986; Bar-Joseph et al., 1983; Enebak et al., 1994; Herrero et al., 2009; Khalifa and Pearson, 2014; Kim et al., 2005; Okada et al. 2011; Rott and Jelkmann, 2001; Jelkmann et al., 1989; Valverde and Sabanadzovic, 2009; Valverde and Sabanadzovic; Valverde et al. 1986; 1990; 2011; Zhang and Rowhani, 2000). Virus specific primers have been developed for RT-PCR detection of most plant and fungal viruses reported to date. After heat denaturation, purified viral dsRNA has been used extensively as the template for reverse transcription PCR (RT-PCR) in plant virus testing (Rott and Jelkmann, 2001; Sabanadzovic and Valverde, 2011; Valverde and Sabanadzovic, 2009; Valverde et al., 2011; Tzanetakis et al 2005). Winter et al. (1992) and Hoyer et al. (1996) characterized Sweet potato chlorotic stunt virus using dsRNA as reagent for RT-PCR, cloning, and labeling. Zhang and Rowhani (2000) developed a rapid cDNA cloning method from dsRNA templates to partially characterized grape viruses. Random cDNA clones from CTV were obtained using denatured viral dsRNA purified from citrus bark tissue (Albiach-Marti et al., 2010). Molecular cloning of plant viruses has been accomplished from small amounts of viral dsRNA after gel purification (Jelkmann et al., 1989). Using dsRNA extracted using the method of De Paulo and Powell (1995), Kunta et al. (2007) conducted RTPCR and were able to detect, clone, and sequence full-length viroids in citrus. Similarly, De la Torre et al. (2009; 2015) detected viroids in peach and avocado using dsRNA extracted by the method of Valverde et al., (1990) followed by RT-PCR. This approach has been particularly useful, when testing plants species known to yield low quality ssRNA.
DsRNA extraction and purification has been suggested as an approach to increase viral sequences in sample preparation for NGS (Wu et al., 2015). In a metagenomics study of viruses in environmental samples, Roossinck (2012) used dsRNA-enriched samples and detected high levels of persistent viruses in plants. De novo assembly of RNAseq results has been accomplished to identify new species of endornaviruses and mycoviruses (Espach et al., 2012; Jo et al., 2015; Khalifa et al., 2016). Using viral dsRNAs, in combination with deep sequencing, researchers have been able to obtain complete genomic sequences of plant and fungal viruses (Al Rwahnih et al., 2011; Candresse et al., 2013; Coetzee et al., 2010; Deker and Parker, 2014; Espach et al., 2012; Quito-Avila et al., 2011; Magae, 2012) and to identify virus-like elements in aquatic microbial populations (Nerva et al., 2016). Deep sequencing from a single grapevine revealed a virome dominated by mycoviruses (Al Rwahnih et al., 2011).
Field surveys
Surveys of RNA viruses in plants and fungi have been conducted by extracting dsRNAs, analyzing them in agarose or polyacrylamide gels, and using them for RT-PCR and sequencing. DsRNAs, isolated from 44 vines from a diseased South African vineyard, were used in a deep sequencing analysis to build a census of the viral population (Al Rwahnih et al., 2011). In a survey for viruses from fungi associated with a parasitic plant (Cuscuta cuspidata) and its host plant (Ambrosia psilostachya) in a tallgrass prairie habitat in Oklahoma, researchers extracted dsRNA to detect fungal viruses (Feldman et al., 2012). The detected viruses were further characterized using reverse transcription-PCR and sequence analysis. A survey for endornaviruses in wild plant species of South Louisiana was conducted using a combination of dsRNA extraction and RT-PCR (Rodrigues de Souto et al., 2015). Of 140 species belonging to 58 plant families, seven were found infected with putative endornaviruses. In Mexico, Piedra et al., (2005) conducted surveys to identify viruses infecting a weed Leonotis nepetifolia (Lamiaceae) commonly present near cultivated fields. Using dsRNA extraction as a complementary tool, the authors were able to identify infections of Alfalfa mosaic virus (AMV), CMV, satellite RNA of CMV, and TMV in this plant species. In a survey for viruses infecting tree tobacco in Southern California, electrophoretic analysis of viral dsRNA was used for virus detection (Valverde and Dodds, 1986). Using dsRNA extraction and electrophoretic analysis, Herrero et al., 2009 conducted a survey of mycoviruses in a collection of 103 isolates belonging to 53 different species of endophytic fungi of grasses and detected dsRNA in 12 isolates.
Plant diseases of unknown etiology
There are several examples of economically important plant diseases of unknown etiology in which the causal agent was elucidated using dsRNA. The causal agent of pallidosis, a disease of strawberry of previously unknown etiology, was identified to be a luteovirus by using purified dsRNA (Tzanetakis et al., 2004; 2005). Similarly, a disease of unknown etiology affecting Japanese holly fern and leather leaf fern was determined to be caused by a new plant virus species from dsRNA that was used for cloning and sequencing the virus which was later named Japanese holly fern mottle virus (Valverde and Sabanadzovic, 2009). Lettuce big vein disease was first shown to be caused by a virus by detection of dsRNAs (Mirkov and Dodds, 1985). In Mexico, the causal agent of a disease of husk tomato (Physalis ixocarpa) which consists of foliar yellow mottle was unknown until 2003 when it was confirmed to be AMV by dsRNA analyses (De la Torre et al., 2003).
Persistent plant viruses
Persistent plant viruses, which are often present in asymptomatic plants, have dsRNA banding profiles that could be confused with that of disease causing acute viruses (Roossinck, 2010; Valverde and Dodds, 1986; Valverde et al., 1990). Persistent plant viruses reported to date belong to the families Amalgaviridae, Chrysoviridae, Endornaviridae, Totiviridae, and Partitiviridae, which include members that infect plants and fungi. There is no evidence that these viruses cause disease in plants. The molecular characterization of most persistent plant viruses has been accomplished by starting with genomic or RF dsRNA purification (Fukuhara, 1999; Pfeiffer, 1998; Okada, et al., 2011; 2013; Sabanadzovic and Valverde, 2011; Sabanadzovic et al., 2016; Sabanadzovic et al., 2010; Sabanadzovic et al., 2009). Using a combination of electrophoretic analysis of viral dsRNA and RT-PCR, Khankhum et al. (2015) demonstrated that two endornaviruses showed differential infection patterns between gene pools of common bean (Phaseolus vulgaris). The presence of persistent viruses in some crop cultivars and wild plant species emphasizes the importance of including samples from symptomless (assumed to be healthy) plants when using the dsRNA technique to detect and identify acute viruses. The dsRNA profiles of three persistent viruses and nucleic acid extracts from two healthy plants are shown in Figure 6.
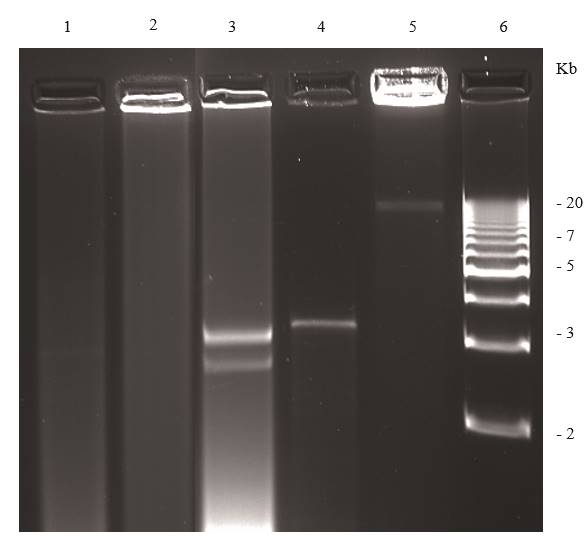
Figure 6. Agarose (1.2 %) gel electrophoresis of dsRNAs extracted from symptomless plants. Lane 1, healthy Phaseolus vulgaris; lane 2, healthy Capsicum annuum; lanes 3-5 plants infected with persistent viruses; lane 3, Persea americana infected with a putative chrysovirus; lane 4, Solanum lycopersicum infected with Southern tomato virus; lane 5 Capsicum annuum infected with Bell pepper endornavirus lane 6 DNA ladder.
Conclusions
Although the cellulose-based protocol for the extraction and purification of viral dsRNA from plants and fungi infected with RNA viruses has been around for over 40 years, it still provides diagnosticians and researchers with a practical tool for the study, detection, and identification of viruses. The simplicity and versatility of the technique, combined with modifications that have improved dsRNA yield, quality, and lowered the overall costs, makes it practical for laboratories with limited resources. As pointed out before, the earlier use of this technique was on the electrophoretic analysis of viral dsRNAs and the use of the banding patterns (electrophoretic profiles) to detect, and in some cases, identify the viruses. However, today, the emphasis is more on the use of viral dsRNA for RT-PCR, cloning, and sequencing which leads to virus identification. It is possible that methods for the extraction of viral dsRNAs will have a place in future implementation of NGS for virus detection and identification. This has been demonstrated with the use of dsRNAs to conduct the complete sequencing of viral RNA genomes from plants and fungi and to identify virus-like elements in aquatic microbial populations using NGS (Nerva et al., 2016), deep sequencing analysis of viruses infecting grapevines (Al Rwahnih et al., 2011; Coetzee et al., 2010), and a metagenomics study of environmental samples (Roossinck, 2012).

