










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista mexicana de fitopatología
On-line version ISSN 2007-8080Print version ISSN 0185-3309
Rev. mex. fitopatol vol.28 n.1 Texcoco Jan. 2010
Artículos científicos
Análisis de Pseudomonas Fitopatógenas Usando Métodos Inteligentes de Aprendizaje: Un Enfoque General Sobre Taxonomía y Análisis de Ácidos Grasos Dentro del Género Pseudomonas
Analysis of Plant–Pathogenic Pseudomonas Species Using Intelligent Learning Methods: A General Focus on Taxonomy and Fatty Acid Analysis Within the Genus Pseudomonas
Bram Slabbinck1, Bernard De Baets1, Peter Dawyndt2 y Paul De Vos3
1 Research Group Knowledge–based Systems, Department of Applied Mathematics, Biometrics and Process Control, Ghent University, Coupure links 653, 9000 Ghent, Belgium.
2 Department of Applied Mathematics and Computer Science, Ghent University, Krijgslaan 281 S9, 9000 Ghent, Belgium.
3 Research Group Knowledge–based Systems, Department of Applied Mathematics, Biometrics and Process Control, Ghent University, Coupure links 653, 9000 Ghent, Belgium.
Recibido: Septiembre 20, 2009
Aceptado Enero 12, 2010
Resumen
La identificación de bacterias fitopatógenas es de alta relavancia. En este trabajo se evaluó la identificación de especies fitopatógenas dentro del género Pseudomonas mediante análisis de esteres metílicos de ácidos grasos (FAME). A partir de una base de datos de FAME, se han generado conjuntos de conjuntos de datos de alta calidad. Dos aspectos fueron investigados: la separación de especies fitopatógenas de Pseudomonas, y la diferenciación del grupo de espcies fitopatógenas de Pseudomonas de las no fitopatógenas. En la primera fase se realizó un análisis de componentes principales para evaluar la variabilidad de los datos. Posteriormente el método de aprendizaje árboles aleatorios fue evaluado para propósitos de identificación. El método inteligente permite aprender de la variabilidad y los patrones de los datos y mejorar la identificación de especies. El análisis de componente principal de especies fitopatógenas mostró claramente sobreposición de grupos de datos. Se desarrolló un modelo de árboles aleatorios que permitió alcanzar una eficiencia de identificación de especies del 71.1%. Discriminar el grupo de especies fitopatógenas del grupo de especies no fitopatógenas fue más sencillo, dado el desempeño de los bosques al azar del 85.9%. Por otra parte se demostró que existe una relación estadística entre los perfiles de ácidos grasos y la patogénesis sobre la planta.
Palabras clave: Diagnóstico, bacterias no patogénicas.
Abstract
The identification of plant–pathogenic bacteria is often of high importance. In this paper, we evaluate the identification of plant–pathogenic species within the genus Pseudomonas by fatty acid methyl ester (FAME) analysis. Starting from a FAME database, high quality data sets were generated. Two research questions were investigated: can plant–pathogenic Pseudomonas species be discriminated from each other and can the group of plant–pathogenic Pseudomonas species be distinguished from the group of non–plant–pathogenic Pseudomonas species. In a first stage, a principal component analysis was performed to evaluate the variability within the data. Secondly, the machine learning method Random Forests was evaluated for identification purposes. This intelligent method allows to learn from the variability and patterns in the data and to improve the species identification. The principal component analysis of plant–pathogenic species clearly showed overlapping data clouds. A Random Forests model was developed that achieved a species identification performance of 71.1%. Discriminating the group of plant–pathogenic plant–pathogenic species from the group of non–plant–pathogenic species was more straightforward, given by the Random Forests identification performance of 85.9%. Moreover, it was shown that a statistical relation exists between the fatty acid profiles and plant pathogenesis.
Key words: Diagnosis, non–pathogenic bacteria.
El género Pseudomonas está clasificado en el dominio Bacteria, phylum Proteob acteria, clase Gammaproteobacteria, orden Pseudomonadales y familia Pseudomonaceae (Brenner et al., 2005). La especie tipo del género es Pseudomonas aeruginosa, la cual fue originalmente descubierta por Schroeter en 1872. El género, sin embargo fue propuesto por Migula en 1894. En marzo de 2008, 95 especies de Pseudomonas fueron válidamente publicadas. Los miembros del género Pseudomonas son bastones típicamente rectos o ligeramente curvados con flagelos polares, presentan un mecanismo aeróbico (respiratorio), pero ninguna especie es fermentativa. La mayoría de especies no crecen en condiciones ácidas (pH menor de 4.5) y son habitantes naturales del agua o suelo. Se han establecido diferentes subgrupos dentro de este género basado en las características fenotípicas o patogénicas. Anteriormente, el agrupamiento pudo estar basado en la producción de pigmentos fluorescentes bajo radiación UV (e.g. P. fluorescens) posteriormente se formaron grupos de especies patogénicas y no patogénicas. Dos ejemplos son P. aeruginosa, un patógeno oportunista de humanos, mientras que P. syringae es un patógeno de plantas (Palleroni, 2005; 2008). Los pioneros en la clasificación taxonómica del género Pseudomonas fueron Palleroni y colaboradores (Universidad de California, Berkeley, USA) quien en 1963 describió cinco grupos de Pseudomonas basados en la hibridación rRNA–DNA. Desde entonces, la taxonomía del género Pseudomonas ha sufrido una serie de re–arreglos y hoy en día solamente el grupo I de rRNA corresponde al género Pseudomonas. Esto implica que un gran número de especies previamente asignadas al género Pseudomonas sensu lato (a menudo referida como las "Pseudomonas") fueron transferidos a rangos genéricos o supra genéricos, principalmente pertenecientes a las clases: Alfa, Beta y Gammaproteobacteria. Ejemplos: Acidovorax, Aminobacter, Brevundimonas, Burkholderia, Comamonas, Halomonas, Methylobacterium, Ralstonia, Sphingomonas, Xanthomonas, etc., citado por Kersters et al., (1996) y Palleroni (2005). En 1996, Moore et al., diferenciaron 2 grupos intergenéricos basados en la secuencia del gen 16S rRNA del grupo de Pseudomonas sensu stricto (= el presente género Pseudomonas): el grupo denominado P. aeruginosa y el otro grupo P. fluorescens cada uno con especies provenientes de diferentes linajes. La mayoría de linajes fueron también agrupados con el análisis FAME por Vancanneyt et al., (1996). En el año 2000, Anzai et al., re–evaluaron 128 especies válidas y no válidas de Pseudomonas basados en datos de la secuencia 16S rRNA y varias especies fueron reasignadas a otros géneros. La complejidad de la taxonomía del presente género Pseudomonas está también demostrada por los análisis del gen rpoB realizado por Tayeb et al., (2005), pero la validación del agrupamiento del rpoB aún necesita datos de la hibridación DNA–DNA (DDH) y de la realización de extensos análisis fenotípicos antes de que se puedan proponer cambios a nivel de especie. Un árbol filogenético construido con el método de máxima verosimilitud del género Pseudomonas basado en la secuencia del gen 16S rRNA se puede observar en la Figura 1. Este árbol incluye las especies bacterianas válidamente publicadas en marzo del 2008. Un gran número de patovares relacionados a patogenicidad en plantas, se han descrito dentro de la especie P. syringae y especies relacionadas. Un estudio de DDH mostró la existencia de nueve discretas genomoespecies (Gardan et al., 1999). En este estudio, nosotros seguimos esta clasificación de genomoespecies como si ellos fueran especies válidamente descritas. El género Pseudomonas pertenece a las bacterias Gram–negativas. Esto implica que las investigaciones iniciales en la composición de ácidos grasos en este género estuvieron enfocadas en la capa de lipopolisacáridos correspondiente (LPS). Esta capa es responsable de una importante fracción discriminatoria de hidroxi–ácidos grasos. Varios investigadores mostraron inicialmente que la mayor fracción de ácidos grasos de la capa de LPS de Pseudomonas aeruginosa estuvo constituida por hidroxi ácidos (Fensom y Grey, 1969; Hancock et al., 1970). En los años 1970, la principal investigación en el contenido de ácidos grasos de los miembros del género Pseudomonas sensu lato fue llevada a cabo en los laboratorios de Moss (Centro de Control de enfermedades, Atlanta, USA) (Dees and Moss, 1975; Dees et al., 1979; Moss et al., 1972; Moss, 1981). Ellos encontraron que los patrones de Esteres matílicos de ácidos grasos (FAME) fueron usados rápidamente para diferenciar especies de Pseudomonas y grupos de especies, debido a que los análisis repetidos de FAME dieron patrones similares. Junto con estas investigaciones sobre el contenido de ácidos grasos bacteriales, se llevaron a cabo investigaciones para el mejoramiento del método analítico del contenido de GC. Por supuesto, otros microbiólogos llevaron a cabo investigaciones sobre el contenido celular de ácidos grasos de las Pseudomonas. Ikemoto et al., (1978) concluyeron que la presencia de ácidos hidroxi, cyclopropano ácidos y ca ácidos de cadenas ramificadas fue característico para los grupos y especies de este género. Interesantemente, estos autores al igual que los primeros investigadores usaron la longitud de la cadena equivalente (ECL) para los picos de detección de FAME. Los valores de ECL se determinaron del logaritmo del tiempo de retención de la cadena recta saturada FAME contra el número de carbonos. Uno de los principales estudios sobre los ácidos 3–hidroxi fue reportado por Oyaizu (1983). A inicios de los 90', las primeras evaluaciones de los sistemas de evaluación comercial para la identificación bacterial basada en FAME, Sherlock MIS (MIDI Inc. Newark, DE, USA), se llevaron a cabo también con esp ecies del género Pseudomonas (Hosterhout et al., 1991; Stead, 1992; Stead et al., 1992). Aquí, el grupo de Stead se enfocó en las especies de Pseudomonas fitopatógenas. Este artículo posteriormente puede estar relacionado con una de las primeras revisiones importantes que se hicieron con FAME donde se analizaron 38, de las 86 especies de Pseudomonas válidamente descritas. Seis grupos de cepas fueron discriminadas basadas principalmente en tres tipos de ácidos grasos hidroxi (también core hidroxi ácidos grasos: 2–hidroxi, 3–hidroxi e iso–ramificado 3–hidroxi), a pesar de que ellos tuvieron menos del 10% del área total de picos. Diferencias cuantitativas en ácidos grasos no hidroxi permitieron diferenciar entre taxa dentro de estos grupos y se encontraron pocas diferencias cualitativas entre los perfiles de taxa incluidas en el mismo subgrupo. Incluso únicos perfiles se encontraron para taxa infraespecíficas (subespecies, biovares, patovares) (Stead et al., 1992). Importantemente, Stead et al., (1992) también encontraron buenas correlaciones entre los agrupamientosrealizados con datos de ácidos grasos y los agrupamientos basados en los resultados de hibridación DNA–DNA y DNA–rRNA. Cuatro años después Vancanneyt et al., (1996) realizaron una evaluación taxonómica de las Pseudomonas, en este estudio se incluyeron 30 especies de Pseudomonas. Nuevamente la presencia de ácidos grasos hidroxi mostró ser un buen marcador para la delimitación de especies y una buena correlación se encontró entre los principales grupos obtenidos del análisis FAME de toda la célula y los grupos basados en hibridación DNA–rRNA. Sin embargo, Vancanneyt et al., (1996) también concluyeron que, el contenido de ácidos grasos de las principales especies, no permite discriminar especies dentro de los diferentes grupos, de estos dos últimos estudios, también se concluyó que los ácidos grasos predominantes en el género Pseudomonas son C16:0, C18:1 y derivados (Stead et al., 1992; Vancanneyt et al., 1996). Sin embargo se considera que los ácidos grasos hidroxi, C10:0 3–hidroxi, C12:0 3–hidroxi y C12:0 2–hidroxi son predominantes (Palleroni, 2005). Para información mas detallada relacionada al contenido FAME de las diferentes especies de Pseudomonas, nosotros nos basamos en las descripciones de las especies y en el Manual Bergey de Bacteriología sietemática (Palleroni, 2005). Con la disponibilidad de un gran número de datos FAME para el género Pseudomonas y de un gran número de especies fitopatógenos de este género, nosotros hemos direccionado nuestra investigación al uso de métodos de aprendizaje inteligente para una identificación global o de las especies a nivel de género con datos FAME (Slabbink et al., 2008; Slabbink et al., 2009) a través de la relación entre la composición bacterial de FAME y la patogénesis en plantas. Sin embargo, en este trabajo, nosotros analizamos la habilidad del análisis FAME para diferenciar entre especies fitopatógenas de Pseudomonas y para distinguir estas especies del grupo de especies de Pseudomonas no patogénicas. Para entender e interpretar la presente investigación y los resultados correctamente, nosotros primero introducimos nuestros datos, el análisis de componentes principales, el uso de técnicas de aprendizaje inteligente y el subsecuente análisis estadístico. Finalmente, presentamos los resultados obtenidos en esta investigación de FAME en especies de Pseudomonas fitopatógenas.
MATERIALES Y MÉTODOS.
Análisis FAME y grupo de datos. Se consideraron las bases de datos FAME del Laboratorio de Microbiología (Ghent University, Belgium) y el BCCM TM/Colección de Bacterias LMG (Bélgica). El análisis FAME de todas las células bacterianas se ha llevado a cabo en este laboratorio desde 1989 y ha resultado en una base de datos que actualmente contiene más de 71,000 perfiles FAME. La investigación basada en FAME está dirigida a bacterias con impacto ambiental, clínico e industrial, pero también se lleva a cabo con propósitos taxonómicos. Básicamente, los perfiles FAME obtenidos por cromatografía de gases son generados durante el crecimiento bacterial, como se describe en el protocolo TSBA del Sistema de Identificación Microbial Sherlock de MIDI Inc. (Sherlock Microbial Identification System de MIDI Inc.) (Newarl, DE, USA). Este protocolo define un estándar para el crecimiento, cultivo y análisis de cepas bacterianas y la reproducibilidad e interpretabilidad de los perfiles es solamente posible trabajando bajo las condiciones descritas. En este trabajo nosotros nos enfocamos en las especies del género Pseudomonas las cuales sobrepuesta del tercer y cuarto cuadrante de la placa estriada. Posteriormente, los FAME se extraen mediante un procedimiento de cuatro pasos: saponificación, metilación, extracción y limpieza de la muestra, finalmente son analizadas por cromatografía de gases. Seguido al análisis por cromatografía de gases, con la ayuda de una calibración y una lista de nombres, los perfiles FAME resultantes son estandarizados mediante el cálculo de las áreas relativas de los picos para cada pico identificado con respecto al área total de los picos. En este trabajo, se usó el cuadro de picos identificados en TSBA. El perfil FAME resultante de toda la célula bacteriana analizado por cromatografía de gases en condiciones de crecimiento estándar es considerado posteriormente como perfiles estándares FAME. En este trabajo se consideraron los datos de las Pseudomonas obtenidos con el análisis FAME por Slabbinck et al., 2009 debido a que se encuentra en concordancia con la taxonomía bacterial de Marzo de 2008. Este grupo de datos cubre 1673 perfiles estándares de FAME de 95 especies de Pseudomonas válidamente publicadas en los que se ha identificado 94 picos FAME. En la presente investigación, este grupo de datos es evaluado en términos de patogénesis en plantas. Aquí, una especie es considerada como fitopatógena cuando una cepa está relacionada a plantas o patogénicas en hongos o setas. En relación a nuestro grupo de datos FAME de Pseudomonas se consideran varias especies de Pseudomonas fitopatógenas. Estas especies están listadas en el Cuadro 1, junto con el número de perfiles estándar de FAME correspondientes, su hospedante (s) relacionado y la referencia (s). En relación a los patovares de Pseudomonas syringae, la clasificación de genomoespecies se realizó de acuerdo a lo sugerido por Gardan et al., (1999).
Aquí, la genomoespecie 3 corresponde a los patovares de P. syringae: antirrhini, apii, berberidis, delphinii, maculicola, passiflorae, persicae, primulae, ribicola, tomato y viburni. La genomoespecie 7 corresponde a los patovares de P. syringae: helianthi y tagetis. Remarcando también que algunas especies de Pseudomonas son generalmente mal nominadas y están transferidas a otros géneros, tales como P. beteli (Stenotrophomonas maltophilia) y P. hibiscola (Stenotrophomonas maltophilia) (Anzai et al., 2000). La especie P. cissicola debería ser transferida al género Xanthomonas, pero se requiriere de un análisis más extensivo de las características genotípicas y fenotípicas de las especies (Anzai et al., 2000; Hu et al., 2007; Palleroni, 2005).
Una anomalía taxonómica similar se observó en P. flectens, la cual basada en el análisis de la secuencia del gen 16S rRNA se agrupó en la familia Enterobacteriaceae (Anzai et al., 2000; Palleroni, 2005). P. beteli, P. cissicola y P. hibiscicola fueron posteriormente anotadas en el grupo P. beteli. Cuando se enfocan en especies de Pseudomonas fitopatógenas, se evalúan dos casos interesantes de pruebas relacionadas con la identificación bacteriana basada en datos FAME: la discriminación de las especies fitopatógenas están listadas en el Cuadro 1 y la discriminación de especies fitopatógenas y no fitopatógenas basadas en datos FAME de las Pseudomonas. En el primer caso, un nuevo grupo de datos se extrajo del grupo de datos completos de FAME cubriendo 25 especies fitopatógenas. En el segundo caso, los perfiles de FAME del grupo completo de Pseudomonas son re–calificadas de acuerdo con las propiedades de infectar plantas u hongos. Así, en este caso, solamente dos etiquetas o clases son considerados: fitopatógenos o no fitopatógenos. Obsérvese que el grupo de las no–fitopatógenas contiene 70 especies de Pseudomonas.
Análisis de componentes principales. Cuando se trata con grupo de datos de una dimensionalidad excesiva (i.e. en este trabajo el número de picos FAME), el enfoque para reducir esta dimensionalidad es combinar las diferentes características y de esta manera, proyectar la gran dimensionalidad de los datos en un espacio dimensional reducido. El análisis de componentes principales (ACP) es un método de reducción de dimensionalidad, en el cual las combinaciones lineares están compuestas de los diferentes picos FAME (también llamado características) (Duda et al., 2001). Inicialmente, la combinación linear que representa la cantidad de variabilidad en los datos es seleccionada y llamado el principal componente principal (PC). Después, las subsecuentes combinaciones lineares son ortogonales al PC anterior, y así repetidamente, estas combinaciones van a representar la varianza más alta. Una visualización simple de los PCA se lleva a cabo dibujando la varianza y la varianza acumulada de los PC, la cantidad de varianza está representada en la gráfica de sedimentación (ver también Figuras 2, (3) y 4). De esta gráfica, es fácil determinar cuántos PC son necesarios para cubrir un cierto porcentaje de la variabilidad en los datos. Estos PC pueden subsecuentemente ser usados para aprender con un número más pequeño de características, resultando en un modelo de bajas dimensiones, o al menos en un modelo menos complejo. Esto no es solamente una ventaja por razones computacionales, sino también puede ser efectivo para incrementar el comportamiento de un modelo de identificación. Graficando los datos en dos o tres espacios dimensionales de componentes principales permite una fácil interpretación de las relaciones (similitud o distancias) entre las diferentes clases, i.e. especies o grupos de especies.


Aprendizaje automatizado.
Terminología. Clasificación e identificación son términos usados con diferentes significados en los campos de la microbiología y en el aprendizaje automatizado. Mientras la identificación se interpreta como la asignación de las clases existentes de un organismo desconocido o datos puntuales, la clasificación debería ser interpretada de manera diferente. En un contexto taxonómico, la clasificación bacterial se refiere a la agrupación de organismos bacterianos basados en similitudes genotípicas y fenotípicas (Madigan et al., 2009). Sin embargo, en el aprendizaje automatizado, el objetivo de la clasificación es describir estadísticamente la relación entre los datos señalados de varias clases, considerando las clases establecidas, por la generalización de las tendencias observadas en los datos (Mitchell, 1997). En el contexto de este trabajo, nos referimos a clasificación como el proceso de construir modelos computacionales para diferenciar entre los perfiles FAME de los diferentes grupos de especies bacterias o grupos de especies (Slabbinck et al., 2009).
Árboles aleatorios. Random Forest (RFs) es un método de agrupación basado en datos contenidos idealmente en una bolsa. Los métodos de agrupación generan múltiples clasificadores (o modelos de clasificación) y los resultados se acumulan en forma de agregados. Específicamente, un RF es una agrupación de árboles para clasificación. En cada nodo de un árbol de clasificación, se elige la mejor división entre un subconjunto de características elegidas al azar en ese nodo. En contraste, los árboles de clasificación estándar dividen a cada nodo en base a todas las características disponibles. Los RF funcionan muy bien comparados con muchos otros clasificadores y es robusto frente a re–muestreos (Breiman, 2001). Para lograr una clasificación óptima de los datos, se requiere un paso de optimización en dos parámetros: el número de características elegidas al azar en cada nodo (Nf) y el número de árboles que se produzcan en el análisis (Nt). La optimización de Nf se realiza variando el número de características de 1 al número total de características en pasos de 5 y la optimización de Nt se realiza variando el número de árboles de 1000 a 4000 en pasos de 250. La optimización de los parámetros se realiza con un filtro de búsqueda y finalmente se elige la combinación de los parámetros que llevan a los más pequeños errores de validación. El primer paso en el algoritmo RF es la generación de muestras Nt bootstrap (muestreo con reemplazo) del conjunto de datos originales. Estas muestras contienen cerca de dos tercios de los perfiles FAME del grupo de datos originales. Los perfiles restantes son usados como un grupo de prueba. Para cada muestra bootstrap, se produce un árbol sin ninguna modificación por el método previamente descrito. Después, para cada muestra, los perfiles de prueba correspondientes son predecibles por el árbol correspondiente. Al final de la corrida de todas las muestras, se asigna un valor a la clase marcada que obtuvo la mayoría de las combinaciones en cada perfil y que estuvo presente en la prueba. La proporción de los errores de clasificación promedio de todos los perfiles probados se toma como la tasa de error global. Nuevos datos pueden ser predecidos por la agregación de los árboles Nt y por selección del que tuvo el mayor número de veces que coincidieron (Breiman, 2001). Los Rfs usados en este estudio son generados con el programa "Random Forest".
Validación. Para la optimización de los parámetros y la evaluación del modelo, se realiza una evaluación cruzada con la unión de los resultados de las pruebas (Parker et al., 2007; Varma y Simon, 2006; Witten et al., 2005). En la validación cruzada, el grupo de datos de entrada se dividen en k partes de igual tamaño, generalmente de una forma aleatoria. Para la parte kth, el modelo funciona con la otra parte k–1, mientras el comportamiento es evaluado con la parte respectiva. Este proceso de entrenamiento y validación se realiza por cada parte y la validación cruzada de la estimación del error es igual al promedio de los errores obtenidos en las diferentes veces (Bishop, 2006; Hastie et al., 2009). En general, cinco y 10 validaciones cruzadas son recomendadas para la selección del modelo (Breiman, 1992; Kohavi, 1995). En muchos mundos reales los grupos de datos, o el número de datos por clase varían. Un buen enfoque para tratar con estas desproporciones es realizar una validación cruzada estratificada. Aquí, los diferentes subgrupos son llamados estratificados de modo que, basados en las diferentes clases, tienen aproximadamente las mismas proporciones de puntos de datos como el grupo de datos originales (Kohavi, 1995). Porque RF no origina errores aleatorios (Breiman, 2001), la optimización de parámetros se realiza en el grupo de prueba. Hemos escogido hacer esto porque algunas especies corresponden a un pequeño número de perfiles FAME y el aprendizaje se basa en un número aún más pequeño de perfiles FAME. Sin embargo, una mejor práctica experimental es realizar la optimización de parámetros en un proceso de validación cruzada y usar los grupos de prueba solamente para propósitos de identificación. Finalmente, el agrupamiento es realizado por la validación cruzada de los "folds" (o de las pruebas de los subgrupos), implicando un modelo de desempeño estimado basado en el conjunto completo de datos. Como las especies son consideradas solamente asociadas con un mínimo de cuatro perfiles FAME por especie, se realizan validaciones cruzadas por cuadriplicado.
Evaluación. Para ambas técnicas, la probabilidad estimada es analizada estadísticamente. Para cada perfil, las especies etiquetadas son consideradas con la probabilidad más alta. Cuando se trata con dos clases, un método popular para la evaluación del comportamiento de un clasificador es la construcción de una matriz confusa. Una matriz confusa resume las predicciones al reportar el número de positivos verdaderos (TP o casos positivos que son previstos como positivos), falsos positivos (FP o casos negativos que son predichos como positivos, verdaderos negativos (TN o casos negativos que son predichos como negativos) y falsos negativos (FN o casos positivos que son previstos como negativos).
De esta matriz pueden ser calculadas diferentes mediciones de desempeño tales como sensibilidad (TP/(TP+FN)), precisión (TP/(TP+FP)) y F–score ((2*S*P)/(S+P), con S y P como sensibilidad y precisión respectivamente. nótese que cuando no se obtienen resultados TP y FP, la precisión lleva a un valor igual al infinito. Para el F–score, este caso, junto con el denominador de cero, puede también llevar a un valor de infinito. Sin embargo, cuando, se confrontan con más de dos especies bacterianas, por lo tanto a un problema de múltiples clases, se puede tener una matriz de confusión compuesta. Subsecuentemente, la evaluación puede ser hecha mediante la fragmentación de la matriz de confusión multiclase a una matriz de confusión N dos clases, donde N es el número de clases o especies consideradas. en cada matriz de confusión de dos clases, las especies correspondientes son evaluadas contra todas las demás especies implementadas en el clasificador. en otras palabras, todas las demás especies son consideradas como una especie ficticia. Finalmente, una medida global puede ser calculada promediando los resultados de las diferentes matrices de confusión.
RESULTADOS Y DISCUSIÓN
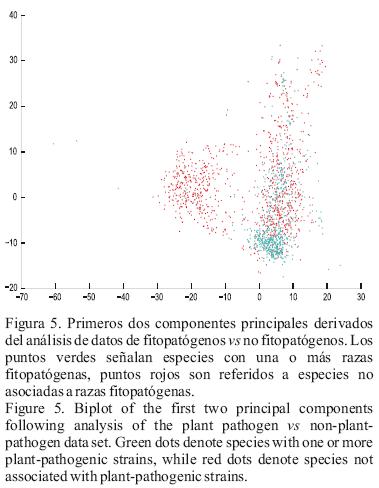
ACP. Los datos que tenemos evaluados hasta ahora de FAME pueden ser usados para discriminar entre especies fitopatogénicas de Pseudomonas, y entre este grupo y especies no fitopatógenas de Pseudomonas. El análisis de sedimentación obtenido corresponde al análisis de componentes principales en ambos grupos de datos. Estos datos están marcados en las Figuras 2 y 4. De ambas figuras, queda claro que tres a cuatro componentes principales pueden representar el 90 a 95% de la variabilidad en los datos. Esto nos indica solamente que muchas de las características (picos) son correlacionados con cada uno, pero también la dimensionalidad del grupo de datos (número de picos) pueden ser significativamente disminuidos, mientras mantenga una discriminación similar posible para las diferentes especies o grupos de especies. Esta correlación puede ser atribuida a la ruta biosintética de ácidos grasos a través de los cuales diferentes ácidos grasos son típicamente convertidos en otras moléculas de ácidos grasos (Madigan et al., 2009). Además, la discriminación entre las diferentes especies o grupos de especies es visualizada por gráficos de los primeros dos componentes principales, ver Figura 3 y 5. Del primer gráfico, el cual corresponde al grupo de datos de fitopatógenos, es claro que las especies fitopatógenas son fuertes para distinguir de otros basados en los datos FAME. También, algunos grupos distintivos pueden inmediatamente ser vistos tales como P. agarici, P. corrugata, P. flavescens, P. tolaasii. Estas especies presentes en el agrupamiento de la izquierda corresponde al grupo de P. syringae (Anzai et al., 2000). Se remarca que en esta gráfica, las especies pertenecientes al grupo P. beteli y la especie de P. flectens no están incluidos permitiendo su cambio a otras especies. Esto puede ser explicado por la distancia entre estas especies y las especies de Pseudomonas sensu stricto, permitiendo posteriormente un estricto agrupamiento de las especies en el espacio respectivo de componentes principales. Cuando comparamos a una especie no fitopatógenas, el segundo biplot muestra que el perfil de FAME de las fitopatógenas se agrupa principalmente en una nube de dispersión y el grupo de datos de las especies no fitopatógenas en dos distintas nubes de dispersión FAME, con una nube claramente sobrepuesta con las nubes FAME de las fitopatógenas.
Aprendizaje automatizado. La primera columna del Cuadro 2 resume los resultados del experimento de aprendizaje automatizado por los cuales se investiga la posible discriminación de las especies de Pseudomonas fitopatógenas. Estos resultados son similares a los resultados del análisis de aprendizaje automatizado de los grupos de datos completos, en el sentido de que son encontrados similares los valores métricos (Slabbnick et al., 2009). Aunque un número más pequeño de especies es investigado, queda claro que no es sencillo para diferenciar las diferentes especies fitopatógenas de otras Pseudomonas. Estos resultados son soportados por los análisis de componentes principales en la sección previa y Figura 1. De los correspondientes gráficos de los primeros dos componentes principales, es obvio que los patrones FAME se sobreponen en las diferentes especies, haciendo que la técnica de aprendizaje automatizado no sea fácil para encontrar una buena función matemática flexible que represente la amplitud de las especies.Podemos concluir que el análisis FAME no es una buena técnica para diferenciar entre todas las especies fitopatógenas de Pseudomonas. Sin embargo, algunas especies podrían ser claramente diferenciadas.

Especies con un F–score más grande que 0.8 son P. cissicola, P. corrugata, P. flavescens, P. flectens, P. fuscovaginae, P. marginalis, P. tolaasii, P. tremae y P. viridiflava. Para algunas de estas especies, grupos distintos podrían también ser encontrados en la gráfica del PCA. Si esta conclusión permanece para especies fitopatógenas en otros géneros este es un tópico para investigaciones futuras. En un segundo experimentos sobre aprendizaje automatizado, se ha investigado como un grupo de especies fitopatógenas de Pseudomonas podrían ser discriminados de los grupos de especies no fitopatógenas. Se estableció el mismo sistema de aprendizaje automatizado usado anteriormente. Sin embargo, debido al gran número de datos disponibles en los diagramas de dispersión disponibles, se decidió llevar a cabo la validación de "ten–fold cross–validation". Nuevamente, debido a un no sobre ajuste de RF, la optimización de parámetros es también hecho por la "cross–validation fold". Los resultados de la prueba de esta segunda clase de experimento para clasificación son reportados en la segunda columna del Cuadro 2. Se puede concluir inmediatamente que es posible una alta discriminación entre especies fitopatógenas y no fitopatógenas usando datos FAME. El alto valor métrico es algo que sorpresivamente se observa con las nubes de dispersión sobrepuestas visualizado en el análisis de componentes principales (ver Figura 5).
Está claro que existen algunas relaciones entre el contenido de ácidos grasos de especies fitopatógenas. La probabilidad estimada obtenida en el experimento RF son estadísticamenteanalizadas con la prueba de Wilcoxon Rank–sum. Esta prueba asume observaciones de muestras al azar que son independientes y continuas y que la forma de la distribuciones subsecuentes son idénticas (Higging, 2004; Sheskin, 2004).En este experimento, los primeros supuestos están confirmados, mientras el último supuesto se considera como presente. Un valor–p de aproximadamente cero es obtenido, implicando probabilidades estimadas estadísticamente diferentes en el nivel de significancia de 0.05. Una posible explicación para esta fuerte identificación podría ser encontrado en el artículo de Stead (1992), quien describe un agrupamiento de un gran número de especies fitopatógenas de Pseudomonas por hidroxi–ácidos grasos. Aunque, en este estudio, se ha considerado principalmente datos del género Pseudomonas sensu lato para discusión. Aquí, un grupo principal corresponde al género Pseudomonas sensu stricto. Este grupo consistió de cerca de 35 taxa de fitopatógenas que podrían ser principalmente discriminadas con base en los ácidos grasos hidroxy C10:0 3–hidroxy y C12:0 3–hidroxy. Subgrupos podrían ser también obtenidos por estos ácidos grasos hidroxy, juntos con el ácidos grasos C12:0 2–hidroxy. Para este grupo, Stead (1992) concluyó que se encontraron muchas diferencias cuantitativas y cualitativas en estos ácidos grasos. No se realizaron comparaciones con especies no fitopatógenas de Pseudomonas. Sin embargo, estas discriminaciones pueden ser también asumidas y es muy valorable para la discriminación entre especies patogénicas y no patogénicas. Futuros estudios de esta relación debería, sin embargo, revelar todos los constituyentes que determina FAME y las diferencias cualitativas y cuantitativas correspondientes.
CONCLUSIONES
Hemos evaluados las posibilidades del análisis FAME para la identificación de especies fitpatogénicas. Se han considerado dos casos; identificación a nivel de especie y una discriminación del grupo de especies fitpopatógenas de las no fitopatógenas. Un simple análisis inicial de los datos fue hecho por el análisis de componentes principales. De estos experimentos, surge claramante que solamente unos pocos componentes principales explican la gran variabilidad en los datos. Esto es principalmente debido a la correlación entre diferentes ácidos grasos. Sin embargo, cuando se observa el biplot del primero de los dos componentes principales, los perfiles de ácidos grasos de diferentes especies fitopatógenas están claramente sobrepuestos. En relación al grupo de datos de los patógenos de plantas versus no patogénicos, una distinta nube de dispersión de los datos puede ser vista. Sin embargo, una sobreposición es aún vista en los datos. Los resultados del análisis de datos es un conocimiento importante cuando se considera el aprendizaje automatizado para propósitos de identificación. Debido a los diagramas de dispersión FAME sobrepuestos, se necesita conocer más de la amplitud de especies flexibles. En el caso de de distinguir entre especies de Pseudomonas fitopatógenas, este proceso de aprendizaje parece ser muy difícil cuando solamente se lleva a cabo una identificación moderada. Sin embargo, algunas especies podrían ser claramente identificadas. Cuando discriminamos las especies fitopatógenas de Pseudomonas, los RFs son capaces de llevar a cabo una buena identificación de los grupos. Además de esto, tenemos estadísticamente también mostrado que los perfiles de FAME de ambos grupos son significantemente diferentes. En otras palabras, una clara relación estadística existe entre ciertos ácidos grasos y lapatogénesis en plantas. Trabajos futuros deberán revelar cual constituyentes juegan un rol principal en esta relación. Otro tópico interesante para futuras investigaciones puede comprender las relaciones entre fitopatógenos en otros géneros bacteriales.
LITERATURA CITADA
Anzai, Y., Kim, H., Park, J.–Y., Wakabayashi, H., and Oyaizu, H. 2000. Phylogenetic affiliation of the Pseudomonads based on 16S rRNA sequence. International Journal of Systematic and Evolutionary Microbiology 50: 1563–1589. [ Links ]
Bishop, C.M. 2006. Pattern Recognition and Machine Learning, 1st Edition, Springer, New York. 738 p. [ Links ]
Breiman, L., Spector, P. 1992. Submodel selection and evaluation in regression: the X–random case. International Statistical Review 60: 291–319. [ Links ]
Breiman,L. 2001. Random Forests. Machine Learning 45: 5–32. [ Links ]
Brenner, D.J., Krieg, N.R., and Staley, J.T. 2005. Bergey's Manual of Systematic Bacteriology Volume 2: The Proteobacteria Part A Introductory Assays, Springer, New York, NY, USA. 304 p. [ Links ]
Catara, V., Sutra, L., Morineau, A., Achouak, W., Christen, R., and Gardan, L. 2002. Phenotypic and genomic evidence for the revision of Pseudomonas corrugata and proposal of Pseudomonas mediterranea sp. nov. International Journal of Systematic and Evolutionary Microbiology 52:1749–1758. [ Links ]
Dees, S.B., Moss, C.W. 1975. Cellular fatty acids of Alcaligenes and Pseudomonas species isolated from clinical specimens. Journal of Clinical Microbiology: 1:414–419. [ Links ]
Dees,S.B., Moss, C.W., Weaver, R.D., and Hollis D. 1979. Cellular fatty acid composition of Pseudomonas paucimobilis and groups IIk–2, Ve–1, and Ve–2. Journal of Clinical Microbiology 10: 206–209. [ Links ]
Duda, R.O., Hart, P.E., and Stork, D.G. 2001. Pattern Classification, John Wiley & Sons, New York, USA. 654 p. [ Links ]
Fensom, A.H., and Gray, G.W. 1969. The chemical composition of the lipopolysaccharide of Pseudomonas aeruginosa. Biochemical Journal 114: 185–196. [ Links ]
Gardan, L., Shafik, H., Belouin, S., Broch, R., Grimont, F., and Grimont, P.A.D. 1999. DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Downson 1959). International Journal of Systematic Bacteriology 49: 469–478. [ Links ]
Gardan, L., Bella, P., Meyer, J.M., Christen, R., Rott, P., Achouak, W., and Samson, R., 2002. Pseudomonas salomonii sp. nov., pathogenic on garlic, and Pseudomonas palleroniana sp. nov., isolated on rice. International Journal of Systematic and Evolutionary Microbiology 52: 2056–2074. [ Links ]
Hancock, I.C., Humphreys, G.O., and Meadow, P.M. 1979. Characterisation of the hydroxy acids of Pseudomonas aeruginosa 8602. Biochimica et Biophysica Acta (BBA) 202: 389–391. [ Links ]
Hastie, T., Tibshirani, R., and Friedman J. 2009. The Elements of Statistical Learning: Data Mining, Inference and Prediction, 2nd Edition, Springer–Verlag, New York, USA. 763 p. [ Links ]
Higgings, J.J. 2004. An Introduction to Modern Nonparametric Statistics, Brooks/Cole–Thomson Learning, CA, USA. 366 p. [ Links ]
Hildebrand, D.C., Palleroni, N.J., Hendson, M., Toth, J., and Johnson, J.L. 1994. Pseudomonas flavescens sp. nov., isolated from walnut blight cankers. International Journal of Systematic Bacteriology 44: 410–415. [ Links ]
Höfte, Μ., and De Vos, P. 2007. Plant–pathogenic Pseudomonas species. p. 507–536. In: G.S. S. (Eds.) Plant–associated bacteria, Springer, Dordrecht, The Netherlands. 712 p. [ Links ]
Hu, F.–P., Yount, J.M., Stead, D.E., and Goto, Μ. 1997. Transfer of Pseudomonas cissicola (Takimoto 1939) Burkholder 1948 to the genus Xanthomonas. International Journal of Systematic Bacteriology 47:228–230. [ Links ]
Ikemoto, S., Kuraishi, H., Komagata, K., Azuma, R., Suto T., and Murooka, H. 1978. Cellular fatty acid composition in Pseudomonas species. Journal of General and Applied Microbiology 24 :199–213. [ Links ]
Janse, J.D., Rossi, P., Angelucci, L., Scortichini, Μ., Derks, J.H.J., Akermans, A.D.L. and De Vrijer,R. 1996. P.G. Psallidas, Reclassification of Pseudomonas syringae pv. avellanae as Pseudomonas avellanae spec. nov. the bacterium causing canker of hazelnut (Corylus avellanae L.). Systematic and Applied Microbiology 19:589–595. [ Links ]
Kersters, K., Ludwig, W., Vancanneyt, Μ., De Vos, P., Gillis, Μ., and Schleifer, K.H. 1996. Recent changes in the classification of pseudomonads: an overview. Systematic and Applied Microbiology 19: 465–477. [ Links ]
Kohavi, R. 1995. A study of cross–validation and bootstrap for accuracy estimation and model selection, Proceedings of the Fourteenth International Joint Conference on Artificial Intelligence, Montréal, Canada, 1995. p. 1137–1145. [ Links ]
Letunic, I., and Bork, P. 2007. Interactive Tree Of Life (ITOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128. [ Links ]
Madigan, M.T., Martinko, J.M., Dunlap, P.V., and Clarck, D.P. 2009. Brock Biology of Microorganisms, 12th Edition, Pearson Education Inc., San Francisco, USA. 1061 p. [ Links ]
Mitchell, T.M., 1997. Machine Learning, McGraw–Hill, Boston, USA. 414 p. [ Links ]
Miyajima, K., Tanii, A., and Akita, T. 1983. Pseudomonas fuscovaginae sp. nov., nom. rev. International Journal of Systematic Bacteriology 33:656–657. [ Links ]
Moore,E.R.B., Mau, Μ., Arnscheidt, A., Böttger, E.C., Hutson, R.A., Collins, M.D., Van de Peer, Y., De Wachter, R., and Timmis, K.N. 1996. The determination and comparison of the 16S rRNA gene sequences of species of the genus Pseudomonas (sensu stricto) and estimation of the natural intrageneric relationships. Systematic and Applied Microbiology 19:478–492. [ Links ]
Moss, C.W., Samuels, S.B., and Weaver, R.B., Cellular Fatty Acid Composition of Selected Pseudomonas Species. Applied Microbiology 24:596–598. [ Links ]
Moss, C.W., and Samuels, S.B. 1974. Short–chain acids of Pseudomonas species encountered in clinical specimens. Applied Microbiology 27: 570–574. [ Links ]
Moss, C.W., 1975. Gas–liquid chromatography as an analytical tool in microbiology. Journal of Chromatography 203:337–347. [ Links ]
Moss, C.W., and Dees, S.B. 1975. Identification of microorganisms by gas chromatographic–mass spectrometric analysis of cellular fatty acids. Journal of Chromatography 112:595–604. [ Links ]
Moss, C.W., and Dees, S.B.1976. Cellular fatty acids and metabolic products of Pseudomonas species obtained from clinical specimens. Journal of Clinical Microbiology 4: 492–502. [ Links ]Moss, C.W. 1981. Gas–liquid chromatography as an analytical tool in microbiology. Journal of Chromatography 203:337–347. [ Links ]
Munsch, P., Alatossava, T., Marttinen, N., Meyer, J.M., Christen, R., and Gardan, L. 2002. Pseudomonas costantinii sp. nov., another causal agent of brown blotch disease, isolated from cultivated mushroom sporophores in Finland. International Journal of Systematic and Evolutionary Microbiology 52:1973–1983. [ Links ]
Osterhout,G.J., Shull, V.H., and Dick, J.D. 1991. Identification of clinical isolates of Gram–negative nonfer–mentative bacteria by an automated cellular fatty acid identification system. Journal of Clinical Microbiology 29:1822–1830. [ Links ]
Oyaizu, H., and Komagata, K., 1983. Grouping of Pseudomonas species on the basis of cellular fatty acid composition and the quinone system with special reference to the existence of 3–hydroxy fatty acids. Journal of General and Applied Microbiology 29:17–40. [ Links ]
Palleroni, N.J., Kunisawa, R., Contopoulou, R., Doudoroff, R., 1973. Nucleic acid homologies in the genus Pseudomonas. Systematic and Applied Microbiology 23: 333–339. [ Links ]
Palleroni, N.J., 1984. Genus I. Pseudomonas Migula 1984. p. 141–199. In: N.R. Krieg, J.G. Holt (Eds.), Bergey's Manual of Systematic Bacteriology Volume 1, Williams and Wilkins, Baltimore, USA. 721 p. [ Links ]
Palleroni, N.J., 2005. Genus I. Pseudomonas Migula 1894, 237AL. . p. 322–379. In: D.J. Brenner, N.R. Krieg, J.T. Staley (Eds.), Bergey's Manual of Systematic Bacteriology Volume 2: The Proteobacteria Part B The Gammaproteobacteria, Springer, New York, USA. 1108 p. [ Links ]
Palleroni, N.J. 2008. The Road to the Taxonomy of Pseudomonas. p. 1–18. In: P. Cornelis (Ed.) Pseudomonas: Genomics and Molecular Biology, Caister Academic Press, Norfolk, USA. 244 p . [ Links ]
Parker, J.P., Günter, S., and Bedo, J. 2007. Stratification bias in low signal microarray studies. BMC bioinformatics 8:1–16. [ Links ]
Pruesse, E., Quast, C., Knittel, K., Fuchs, B.M., Ludwig, W.G., Peplies, J., and Glõckner, F.O. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research: 35:7188–7196. [ Links ]
Sheskin, D.J. 2004. Handbook of Parametric and Nonparametric Statistical Procedures, 3rd Edition, Chapman and Hall/CRC, Florida, USA. 1193 p. [ Links ]
Slabbinck, B., De Baets, B., Dawyndt, P., and De Vos P. 2009, Towards large–scale FAME–based bacterial species identification using machine learning techniques. Systematic and Applied Microbiology 32:163–176. [ Links ]
Smith, I.M., Dunez, J., Philips, D.H., Lelliot, E.A., and Archer, S.A. 1988. European Handbook of Plant Diseases, Blackwell Scientific Publications, Oxford, UK. 583 p. [ Links ]
Stamatakis, A. 2006. RAxML–VI–HPC: maximum likelihood–based phylogenetic analysis with thou– sands of taxa and mixed models. Bioinformatics: 22:2688–2690. [ Links ]
Stead, D.E. 1992. Grouping of plant–pathogenic and some other Pseudomonas spp. by using cellular fatty acid profiles. International Journal of Systematic Bacteriology 24:281–295. [ Links ]
Stead, D.E., Sellwood, J.E., Wilson, J., and Viney, I. 1992. Evaluation of a commercial microbial identification system based on fatty acid profiles for rapid, accurate identification of plant–pathogenic bacteria. Journal of Bacteriology 72:315–321. [ Links ]
Tayeb, L.A., Ageron, E., Grimont, F., and Grimont P.A.D. 2005. Molecular phylogeny of the genus Pseudomonas based on rpoB sequences and application for the identification of isolates. Research in Microbiology 156:763–773. [ Links ]
Van Den Mooter, Μ., and Swings, J. 1990. Numerical analysis of 295 phenotypic features of 266 Xanthomonas strains and related strains and an improved taxonomy of the genus. International Journal of Systematic Bacteriology 40: 348–369. [ Links ]
Vancanneyt, Μ., Witt, S., Abraham, W., Kersters, K., and Frederickson, H.L. 1996. Fatty acid content in whole–cell hydrolysates and phospholipid fractions of Pseudomonads: a taxonomic evaluation. Systematic and Applied Microbiology 19:528–540. [ Links ]
Varma, S., and Simon, R. 2006. Bias in error estimation when using cross–validation for model selection. BMC Bioinformatics 7: 91. [ Links ]
Witten, I.H., and Frank, E. 2005. Data Mining Practical Machine Learning Tools and Techniques, 2nd Edition, Morgan Kaufmann, San Francisco, USA. 525 p. [ Links ]

