










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalud Pública de México
versión impresa ISSN 0036-3634
Salud pública Méx vol.53 no.5 Cuernavaca sep./oct. 2011
ARTÍCULO DE REVISÍON
Predisposición genética para el cáncer de mama: genes BRCA1 y BRCA2
Genetic predisposition for breast cancer:
BRCA1 and BRCA2 genes
Steven A Narod, MDI; Adriana A Rodríguez, MD, MScII
IWomen's College Research Institute, University of Toronto.Toronto, Canadá
IILester and Sue Smith Breast Center and Department of Molecular and Cellular Biology, Baylor College of Medicine. Houston, USA
RESUMEN
El descubrimiento de los genes BRCA1 y BRCA2 ha llevado a la introducción de pruebas genéticas cada vez más sofisticadas para medir el riesgo de cáncer de mama de origen hereditario, entre otras cosas. En el presente artículo exploramos los criterios a seguir para realizar pruebas para estos genes, así como las implicaciones en el tratamiento para los pacientes en caso de identificarlos.
Palabras clave: oncogenes; genética; neoplasias de la mama; BRCA1; BRCA2
ABSTRACT
The discovery of genes BRCA1 and BRCA2 has led to the introduction of genetic tests more complex every time for the evaluation ofthehereditarycancerrisk,amongothers.In the present paper we explore the criteria to decide when to run the testing for the genes, as well as the implications for the treatment of patients who are identified with them.
Key words: oncogenes; genetics; breast neoplasms; BRCA1; BRCA2
La etiología del cáncer de mama es desconocida, pero se han implicado factores de riesgos hormonales, reproductivos y hereditarios.1,2 Aunque la mayoría de los cánceres de mama son esporádicos, los avances de la genética han demostrado la base hereditaria para un subgrupo de formas de cáncer.2,3 Actualmente, el cáncer de mama hereditario representa aproximadamente entre 5 y 10% de todos los casos de cáncer de mama.4-6
Se han descrito síndromes de cáncer hereditario donde existen mutaciones en la línea germinal, entre las que se encuentran mutaciones de los genes BRCA1 y BRCA2 en el cáncer hereditario de mama-ovario.2-7
Con el advenimiento de las pruebas genéticas en la práctica oncológica, se logró detectar la susceptibilidad de familias de alto riesgo.5,6,8,9 En la práctica clínica dia-ria, sólo los genes BRCA1 y BRCA2 son indicación para solicitar una prueba genética. Se han encontrado otros genes a los que se les ha adjudicado la particularidad de ser predisponentes para el cáncer de mama, pero no son una premisa para realizar una prueba genética.
Aunque el despistaje genético para el BRCA1 y el BRCA2 no se realiza con frecuencia en la mayoría de los países latinoamericanos, en otros países como Estados Unidos, Canadá, Polonia, Israel y en muchos otros países de Europa Occidental, forma parte de la batería de estudio para evaluar pacientes que potencialmente puedan tener cáncer de mama de origen hereditario.10 Debido a la alta sobrevida del cáncer de mama cuando es tratado en estadios tempranos,8,11 poder identificar a aquellas mujeres que posean un alto riesgo a través de los exámenes genéticos tiene una implicación muy importante en cuanto a salud pública se refiere. Es por esto que el propósito de este artículo es profundizar en el estudio del cáncer de mama hereditario, saber cuándo y a quién se le debe indicar una prueba genética y describir sus características particulares, medidas preventivas, seguimiento, vigilancia y tratamiento.
BRCA1 y BRCA2
El descubrimiento del gen BRCA1 abrió paso a un mejor entendimiento tanto del cáncer de mama hereditario como del cáncer de mama esporádico y llevó a nuevas opciones terapéuticas y preventivas. El gen BRCA1 fue nombrado por primera vez en 1991 por Mary-Claire King. Su grupo lo asignó al cromosoma 17 después de analizar la relación de un gran grupo de familias con casos de cáncer de mama detectado a edades tempranas; pero el paso definitivo fue la identificación de las mutaciones truncadas en el código de secuencia del BRCA1 en familias con múltiples casos de cáncer de mama.1 Algunas familias con alta incidencia de cáncer de mama en hombres resultaron ser no portadoras del BRCA1, lo que llevó a los investigadores a buscar otros genes. Fue entonces en 1994 cuando se relacionó el cromosoma 13 con el gen BRCA2, y un año después fue clonado por el mismo grupo.12 El cáncer de mama en hombres es una alteración rara que representa menos de 1% de todos los cánceres y causa 0.1% de las muertes por cáncer entre los hombres.4,2,12
BRCA1 y BRCA2 son genes supresores de tumores que codifican las proteínas que funcionan en el proceso de reparación del ADN. Por lo tanto, una mutación o una deleción de un gen supresor tumoral provocarían una pérdida de su función y como consecuencia aumentaría la probabilidad de que se desarrolle un tumor. Aunque individuos con el síndrome de cáncer de mama-ovario hereditario heredan un solo alelo defectuoso en BRCA1 o en BRCA2 de su madre o de su padre, tienen un segundo alelo que es funcional. Ahora bien, si este segundo alelo es afectado, se puede desarrollar una célula cancerígena a través de la acumulación de mutaciones adicionales del ADN de la célula.1
Las mutaciones que se reportaron primero en el BRCA1 fueron inserción, deleción intrónica o mutaciones de tipo terminal (mutación "nonsense"). Estas mutaciones usualmente generan una proteína BRCA1 acortada y no funcional.1,2
Las técnicas más comúnmente utilizadas para detección de mutaciones son la de análisis de proteínas truncadas (PTT), la desnaturalización de cromatografía líquida de alto rendimiento (DHPLC), amplificación múltiple de sondas ligando-dependientes (MLPA) y la secuenciación directa de ADN. La técnica del PTT ha dado buenos resultados pero, sin duda, aunque su costo es más elevado, el secuenciamiento completo del gen es la mejor prueba.1
Características del cáncer de mama hereditario
a. Características demográficas:
La prevalencia de las mutaciones BRCA1 y BRCA2 varía de acuerdo con el país y el grupo étnico.6,10,13-15 En Norteamérica se estima que la frecuencia de mutaciones BRCA1 y BRCA2 oscila desde 1 en 250 mujeres a 1 en 800.15,6 Entre los diferentes grupos étnicos la frecuencia más alta se encuentra en los individuos con ancestros judíos asquenazí (1 en 50).15 Otros grupos con alta prevalencia de mutaciones incluyen países como Islandia, Canadá (especialmente franco-canadienses), Polonia y Holanda.6 Estas altas tasas de prevalencia se deben a la presencia de mutaciones fundadoras, que son una o más mutaciones específicas en esa población que han sido heredadas de un ancestro común y que se han ido amplificando a través de las generaciones, contribuyendo a ello el aislamiento geográfico de la población. Por ejemplo, en la población judía asquenazí, existen dos mutaciones fundadoras en BRCA1 (185delAG y 5382insC) y una en BRCA2 (6174delT) que representan más de 90% de las mutaciones observadas en esta población.15 Pero en países o regiones donde los orígenes de la población son mixtos debido a la variedad y a la mezcla de razas, siguen siendo necesarias más pruebas genéticas de secuenciamiento completo en ambos genes para poder determinar con precisión mutaciones fundadoras.6,13
b. Investigación de las mutaciones fundadoras
Para establecer la presencia de una mutación fundadora es necesario identificar la distribución de las mutaciones en una extensa muestra de individuos afectados que no estén emparentados entre sí, confirmarlas a través del secuenciamiento completo y después establecer cuáles mutaciones representan a la mayoría en esa población. La importancia de determinar las mutaciones fundadoras estriba principalmente en la disminución de los costos. Si lográramos disminuir los costos, se podrían ofrecer en una forma más amplia y abarcar un mayor número de mujeres. Debido a que el riesgo de desarrollar cáncer en los portadores de una mutación BRCA1 o BRCA2 es mucho mayor que el de la población general, se llega a la conclusión de que detectar una mutación en una población permitiría establecer medidas preventivas y ofrecer un tratamiento precoz, causando un impacto en cuanto a morbimortalidad se refiere.
El problema de realizar el secuenciamiento completo es que los costos de comercialización son muy elevados y, por lo tanto, se limita la disponibilidad de la prueba y su aplicación a amplia escala. La poca disponibilidad se ve acrecentada por la escasez de centros entrenados y capacitados para practicar estas pruebas genéticas, pues son laboriosas y requieren tiempo.
Por supuesto que no siempre la mutación fundadora identifica las pacientes de alto riesgo; existen excepciones como, por ejemplo, el caso de una mujer que tenga una historia familiar significativa de cáncer de mama y el resultado de la prueba para la mutación fundadora sea negativo. En este caso, es necesario proseguir con la realización de una prueba de secuenciamiento completo.
Otro de los problemas de las pruebas genéticas es aquel que resulta cuando se obtienen mutaciones de tipo terminal (mutaciones que son el resultado del cambio de un solo aminoácido), ya que son más difíciles de interpretar. Se han desarrollado modelos que ayudan a determinar si una mutación que altere sólo un aminoácido residual es deletérea o es una variante inocua, pero no han sido fáciles de incorporar a la práctica clínica debido al gran número de mutaciones terminales identificadas.
Se espera que el costo del secuenciamiento disminuya lo suficiente en un futuro cercano para poder hacer del secuenciamiento completo del gen una alternativa viable en la búsqueda de mutaciones fundadoras.
c. Comportamiento biológico y evolución natural de la enfermedad
Existen cinco subtipos diferentes de tumores: luminal A, luminal B, tipo mamario normal, sobre expresión HER2 y tipo basal, los cuales presentan diferentes comportamientos biológicos. El subtipo basal se asocia con mal pronóstico y es observado en 80 a 90% en las portadoras de mutaciones BRCA1. Los tumores de tipo basal se caracterizan por la expresión de genes (CK5/6/14/17)usualmente encontrados en las células mioepiteliales de la capa basal en la porción ductal terminal de la mama normal.16-19. Se asocian con la negatividad de los receptores de estrógenos, de progesterona y del receptor del factor de crecimiento epidérmico (HER 2 neu), por lo que se denomina cáncer de mama triple-negativo. Se estima que 11% de los cánceres de mama asociados con mutaciones BRCA1 son triple-negativos, se diagnostican a temprana edad y son del tipo basal.19 La proporción de cánceres asociados a mutaciones BRCA1, que son negativos para los receptores de estrógenos, es inversamente proporcional a la edad de la paciente; esto quiere decir que a medida que el cáncer se diagnostica de forma más temprana, es más probable que los receptores estrogénicos resulten negativos. El 81% de los cánceres BRCA1 diagnosticados en pacientes menores de 45 años son negativos para los receptores de estrógenos, mientras que el porcentaje disminuye a 65% en aquellos cánceres diagnosticados después de los 65 años.16,17 En contraste con la alta proporción de cánceres asociados con mutaciones BRCA1 que son negativos para los receptores de estrógenos, sólo 15% de los que son triple negativos están asociados con mutaciones BRCA1.20
En general, los cánceres asociados con mutaciones BRCA1 son ductales infiltrantes, de tipo basal, con un alto grado histológico, con características medulares, infiltración linfocitaria, patrón de crecimiento sincicial y resultan ser triple-negativos. Además se asocian con un comportamiento más agresivo y tienen un peor pronóstico. Esta es la razón por la cual los cánceres de mama que se diagnostican a temprana edad tienen la indicación de realizárseles una prueba genética, mientras que los relacionados al BRCA2 parecen no diferir de los cánceres de origen no hereditario.18,21
En resumen, el fenotipo de los cánceres asociados con las mutaciones del BRCA1 se caracterizan por lo siguiente:16-18
-
Morfológicamente relacionado con cáncer ductal de tipo no especifico (75%) y medular atípico (10%).
-
Generalmente de alto grado histológico (grado III; 75%)
-
Receptores de estrógenos negativos (75%)
-
Expresión del Her2/Neu negativa (95%)
-
Expresión del p53 positiva en 50%
-
Expresión de la ciclina D1 negativa (90%)
-
La presencia de carcinoma in situ es rara
Y el fenotipo de los cánceres asociados con las mutaciones del BRCA2 por lo siguiente:16-18
-
Morfológicamente asociado con el cáncer ductal de tipo no específico (75%), medular atípico (<5%), lobular o ductal con características de lobular más prevalente que en las mujeres con mutaciones BRCA1 (~10%)
-
Grado histológico intermedio (grado II; 45%) a alto (grado III; 45%)
-
Receptor estrogénico positivo (75%)
-
Expresión del Her2/Neu negativa (95%)
-
Expresión del p53 positiva (40%)
-
Expresión de la ciclina D1 positiva (60%)
-
La presencia conjunta de carcinoma in situ es común.
Asesoramiento genético de las familias de alto riesgo
El enfoque de acercamiento a las mujeres que pudieran ser portadoras de una mutación BRCA debe incluir los siguientes planteamientos:
1. ¿A quién se le va a realizar la prueba?
2. ¿Cuáles son los riesgos de contraer cáncer de mama o de ovario?
3. ¿Qué medidas preventivas se pueden utilizar?
4. ¿Cuál es el tratamiento más indicado una vez que el cáncer se ha desarrollado?
Los factores que sugieren que una mutación BRCA1 o BRCA2 pudiera ser encontrada en una familia en particular incluyen el número de parientes afectados con cáncer de mama o con cáncer de ovario, la edad del diagnóstico del cáncer de mama (la edad no influye en caso del cáncer de ovario), tener ancestros judíos, ciertas características anatomo-patológicas de los cánceres de mama y de ovario, y que tengan los receptores triplenegativos.1,3,21
Se ha hecho un gran esfuerzo en la última década por identificar a aquellas familias con historia de cáncer de mama que pudiera ser originado por mutaciones en los genes BRCA. Se han reportado casos de cáncer de mama que ocurren en familias que no son portadoras de una mutación BRCA. Algunos casos puede ocurrir al azar, pero otros podrían deberse a una larga deleción o a la pérdida de la función de mutación del BRCA1 o BRCA2 que no ha podido ser detectada por el tamizaje convencional, o bien porque se debe a mutaciones en otros genes.1,2,4,9
Las pacientes portadoras de una mutación BRCAson consideradas de forma diferente a las pacientes de la población general. A estas pacientes se les ofrecen pruebas de vigilancia más intensas con la idea de realizar una detección del cáncer más temprana, de indicar quimioterapia profiláctica o de practicar mastectomía para reducir el riesgo.1,8,22
¿A quién le vamos a indicar la prueba genética?
Los criterios para indicar una prueba genética varían de acuerdo con el país y con la población estudiada. Encaso de no haber una buena historia familiar, se ofrece hacer una prueba genética a todas aquellas mujeres que tengan cáncer de mama, con los tres receptores negativos y que hayan sido diagnosticadas a los 40 añosde edad o antes.19 Se ofrece también a todas aquellas mujeres con cáncer invasivo de ovario8 y a todas aquellas mujeres judías con cáncer de mama.15 En el caso de que estas tres condiciones no estén presentes, se hace la prueba a las mujeres que tengan una historia familiar significativa: dos o más casos de cáncer de mama diagnosticados a temprana edad (menores de 50 años) o cáncer de ovario diagnosticado a cualquier edad.4,5,23
Recientemente, también se incluyen dentro de los criterios de elección para una prueba genética a las pacientes diagnosticadas con cáncer seroso de ovario de alto grado, cáncer peritoneal primario, cáncer de las trompas de falopio o antecedentes de cáncer de mama en un hombre de la familia, pues estas mujeres tienen una alta probabilidad de tener mutaciones en la línea germinal BRCA1 o BRCA2.1,22,24
Una vez que las pacientes han sido elegidas para realizarles la prueba genética, ésta es asesorada por un médico genetista que documentará un historial de la familia que incluya al menos tres generaciones. Una vez obtenida esta información, el genetista puede entonces calcular el riesgo utilizando diferentes modelos: BRCAPRO, Myriad II, Couch, FHAT, Penn II, Manchester, BOADICEA o IBIS.25 En un estudio reciente se determinó que el modelo BRCAPRO puede ser utilizado en la población hispana con el mismo resultado que en la población de raza blanca.26
Se recomienda el asesoramiento genético antes de indicar la prueba y al tener los resultados. El factor costo-efecto que esto implica debe ser considerado en los países latinoamericanos.
Los factores a considerar para elegir el modelo de asesoramiento del riesgo que se debe usar son: ser altamente sensible, la información debe ser fácil de obtener, el ingreso de la información debe ser fácil y rápido, y debe ser aplicable a la mayoría de los pacientes independientemente del estatus del cáncer, del sexo o de la historia familiar. El asesoramiento genético debe considerarse obligatorio para las pacientes de alto riesgo que son aquellas que poseen un riesgo mayor o igual a 20-25% de heredar la mutación.25
Al momento de evaluar la historia familiar de una paciente es muy importante recordar que el cáncer de mama y de ovario de origen hereditario puede ser transmitido por el padre o por la madre. Por esta razón, la adopción limita la interpretación del historial, al igual que la historia de los parientes con histerectomías u ooforectomías practicadas a temprana edad. Esto puede enmascarar la detección de una predisposición genética en esa familia. Otro factor que pudiera enmascarar una predisposición genética es el hecho de que algunas familias cuentan con pocas hijas, por lo tanto, si en una familia con pocos parientes femeninos se encuentra un caso aislado de cáncer de mama diagnosticado antes de los 50 años, se considera conveniente realizar una prueba genética.7,27 Existen ciertas limitaciones para construir una buena historia familiar: una de ellas es que ciertos parientes se niegan asometerse a las pruebas, ya sea por falta de información, por conflictos familiares o por razones demográficas. La tarea del clínico durante el proceso de asesoramiento genético consiste en ayudar a que el sujeto aclare todas sus dudas, que comprenda el riesgo que posee, el impacto que puede causar en sus familiares, el impacto individual de ser portador y que conozca las alternativas que tiene para evitar o minimizar ese riesgo. 7,27
¿Cuáles son los riesgos de contraer cáncer de mama o de ovario?
El riesgo de cáncer de una portadora puede resumirse de la siguiente forma:
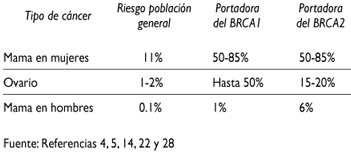
La penetrancia de las mutaciones BRCA sigue siendo materia de investigación. Es necesario invertir esfuerzos en determinar la penetrancia del BRCA1 más que la de cualquier otro gen, ya que la información exacta de los riesgos a la paciente es crucial para que pueda decidir con anticipación qué tipo de medida preventiva va a utilizar. Penetrancia se define como el riesgo de una mujer portadora de desarrollar cáncer de mama o de ovario a lo largo de su vida (usualmente se calcula hasta los 70 años de edad).
Ambos, BRCA1 y BRCA2, tienen valores de penetrancia que oscilan alrededor de 80%.4 El porcentaje de riesgo de una portadora BRCA1 de desarrollar cáncer de ovario es de alrededor de 40%, y el riesgo de una portadora BRCA2 es de 20%, aunque lo desarrollan a una edad más avanzada.23,29,30
La mutación BRCA2 encontrada en mujeres judías (6174delT) tiene una penetrancia menor que la de la mutación 185delAG. Se estima que la penetrancia de la mutación BRCA2 6174delT es de cerca de 28%, mientras que las otras mutaciones BRCA2 están alrededor de 80%.15
También es importante recordar que la penetrancia varía según los países por razones que no están relacionadas con la genética. Por ejemplo, el uso de anticonceptivos orales, la ooforectomía y la paridad tienen influencia en el riesgo de desarrollar cáncer de ovario,21,22,29 Por último, a medida que las mujeres portadoras conocen su estatus genético, la prevalencia del cáncer hereditario debería disminuir, pues seguramente van a tomar medidas preventivas como la ooforectomía y la mastectomía profiláctica.11
Manejo e interpretación de los resultados de las pruebas genéticas
Lo ideal es iniciar las pruebas genéticas en el individuo que se encuentre afectado de cáncer.
Resultado negativo
En la paciente afectada de cáncer: la falta de detección de la mutación en una paciente provee una información limitada y debe ser interpretada con mucho cuidado, pues la causa de cáncer no ha sido aún establecida. Las posibilidades pueden ser que el cáncer esté asociado a una mutación que no ha sido detectable por el método utilizado, que sea causado por una susceptibilidad genética diferente o porque es el resultado de factores no hereditarios.31
En las(os) parientes no afectados: confirma que esta persona no ha heredado la mutación específica para esa familia.
Tanto en el caso de la paciente afectada de cáncer como de la no afectada, la familia debe saber que un resultado negativo no elimina la posibilidad de que exista un factor hereditario en la familia.21,22,31
Resultado positivo
En la paciente afectada de cáncer: esto confirma la asociación del cáncer con un origen genético y preestablece una mutacion específica para esa familia.
En las(os) parientes no afectadas: confiere un riesgo aumentado para los cánceres asociados al BRCA1 o al BRCA2. En este caso se recomienda ofrecer tratamiento profiláctico y seguimiento adecuado.21,22,31
Resultado no concluyente: el resultado puede revelar una variación incierta de significado clínico indeterminado. Generalmente esto se debe a un cambio en un nucleótido simple de ADN que puede o no provocar disfunción en la proteína. Para evaluar este resultado, el laboratorio puede requerir muestras de sangre adicionales de otros miembros de la familia (usualmente aquellos individuos afectados o familiares directos, que son los padres). Estos estudios pueden revelar que la variante es una mutación patogénica o que es un polimorfismo sin significado clínico familiar.21,22,31
¿Qué medidas preventivas se pueden utilizar?
Las estrategias actuales para la prevención primaria del cáncer de mama incluyen mastectomía profiláctica y quimioterapia preventiva con tamoxifeno. La mejor estrategia para reducir el riesgo se obtiene con la mastectomía preventiva (más de 95% de protección.)8,32,33 Sin embargo, esta opción es elegida únicamente por pocas mujeres debido a la preocupación por su imagen corporal y por factores socioculturales.33 Es también importante mencionar que la comunidad médica apoya la decisión de llevar a cabo cirugía preventiva.28 Para facilitar el hecho de que una mujer tome la decisión de realizarse una mastectomía preventiva es necesario que cuente con un adecuado apoyo de su entorno y que también se le ofrezca la opción de realizarse posteriormente una cirugía reconstructiva.32
a. Quimioterapia profiláctica con tamoxifeno. El tratamiento con tamoxifeno está asociado con una reducción en el riesgo de padecer cáncer de mama contralateral de 40 a 70%.33,34 No se sabe hasta dónde el tamoxifeno reduce el riesgo de cáncer de mama primario y, hasta ahora, la utilización del tamoxifeno como quimioterapia preventiva no ha sido aceptada ampliamente.33 Teóricamente, el tamoxifeno no debería reducir la incidencia de cánceres que son receptores estrogénicos negativos, como ocurre en los cánceres BRCA1 asociados.17
b. Cirugía preventiva. Cualquier tratamiento ofrecido alas portadoras tiene que tomar en cuenta el altísimo riesgo de cáncer de mama contralateral y de ovario que pueden tener. Es por esto que a una mujer con cáncer de mama que tenga una mutación BRCA1 o BRCA2 se le debe ofrecer una cirugía más extensa (usualmente mastectomía bilateral), con la finalidad de prevenir un cáncer secundario o un cáncer de mama contralateral.11 Se ha estimado que el riesgo en portadoras del BRCA1 de padecer cáncer de ovario después de 10 años de haber sido diagnosticado el cáncer de mama es de 13%.14
La mastectomía profiláctica bilateral reduce el riesgo de cáncer de mama en más de 90-95%, dependiendo del tipo de mastectomía. La mastectomía total (remueve mama, areola y pezón) es la más efectiva. La mastectomía subcutánea (conserva areola y pezón) deja una mínima cantidad de tejido mamario remanente que en portadoras BRCA siempre implica un riesgo de desarrollar una neoplasia, sin embargo, estos hallazgos no son clínicamente significativos.32,35
La salpingo-ooforectomía profiláctica, cuando se realiza antes de los 45 años de edad, está asociada con una reducción del riesgo de cáncer de ovario de 80%24 y con una reducción del riesgo de cáncer de mama de 50%.36 Los autores de este estudio han demostrado recientemente que la terapia de reposición hormonal pudiera ser ofrecida después de la ooforectomía preventiva para aliviar los síntomas de la menopausia, sin incrementar el riesgo de desarrollar cáncer de mama.24,28,35,37
La salpingo-ooforectomía debería ofrecerse a los 40 años de edad o una vez completada la paridad. Durante la cirugía se deben remover los ovarios completamente y también las trompas de falopio. Se debe realizar una visualización exhaustiva de la pelvis y realizar un lavado peritoneal.24,28
Seguimiento y vigilancia
Tradicionalmente, la pesquisa del cáncer de mama incluye mamografía, autoexamen de la mama y examen clínico de la mama. Actualmente, el Colegio Americano de Obstetricia y Ginecología (ACOG) y la Sociedad de Ginecología Oncológica (SGO) recomienda realizar un examen clínico bianual de la mama, así como también una mamografía anual y una resonancia magnética anual desde los 25 años de edad o antes, según la edad a la que fue diagnosticado el primer caso de cáncer en la familia.28 De acuerdo con una revisión realizada por la USPSTF (U.S. Preventive Services Task Force) las recomendaciones en mujeres de alto riesgo son las siguientes:38
-
Despistaje del cáncer de mama en portadoras BRCA: autoexamen mensual desde los 18 años; examen médico anual o bianual desde los 25-35 años; mamografía anual desde los 25-35 años.
-
Despistaje del cáncer de ovario en portadoras BRCA: ultrasonido transvaginal y CA-125 anual o bianual desde los 25-35 años; es opcional para las portadoras BRCA2.
Recientemente, en un estudio realizado en las Bahamas que está en vías de publicación se propusieron las siguientes pautas para el manejo de pacientes no afectadas de cáncer portadoras de una mutación BRCA1:
1. Mujeres entre 25 y 70 años sin historia de cáncer de mama:
a. Examen mamario mensual practicado por un médico cada 6 meses, desde los 25 años de edad hasta los 70.
b. Mamografía anual desde los 25 años de edad hasta los 70.
c. Si es posible, considerar una resonancia magnética anual.
d. Discutir las opciones que ofrece la mastectomía profiláctica a la edad de 30 años. La cirugía conservadora del pezón puede ser una opción.
e. Si la paciente no desea someterse a una mastectomía profiláctica, entonces se le puede recomendar tomar tamoxifeno 20 mg/día por 5 años. No se debe iniciar este tratamiento antes de los 30 años de edad ni después de los 60.
f. La salpingo-ooforectomía profiláctica (SOB) se recomienda a mujeres entre 35 y 65 años, aunque lo ideal es no más allá de los 40 años. Después dela SOB, ofrecer terapia de reposición hormonal (TRH) con 0.625 mg de estrógenos conjugados. Si el útero está intacto, se puede añadir progesterona. Se puede continuar hasta los 50 años y luego reevaluar. Si la paciente está tomando tamoxifeno, no se recomienda la TRH.
g. Sesiones educativas para las pacientes acerca de los signos del cáncer de mama. En caso de palparse un nódulo, debe inmediatamente ser examinada por un médico. Establecer una forma de contacto fácil y accesible para la paciente.
h. Sesión informativa anual para las mujeres portadoras sobre las actualizaciones relacionadas con los genes BRCA1/2.
2. Mujeres entre 25 y 70 años con historia de cáncer de mama:
a. Se recomienda la mastectomía contralateral antes de los 60 años de edad.
b. Salpingo-ooforectomía entre los 25 y 65 años de edad (ideal antes de los 40).
c. La TRH no está indicada. Si la paciente es sintomática, se recomienda el uso de cremas con estrógenos vía intravaginal.
3. Parientes en riesgo:
a. Dentro de lo posible, se debe aconsejar genéticamente a todos los parientes del sexo femenino mayores de 20 años, y posteriormente ofrecer la prueba genética si el caso lo amerita.
En Estados Unidos y Canadá la resonancia magnética (RM) ha sido añadida al esquema de seguimiento, porque de acuerdo con estudios prospectivos, se ha demostrado consistentemente que la sensibilidad de la RM es mayor que la de cualquier otra modalidad.39 Sin embargo, no ha sido demostrado aún que el uso regular de la resonancia reduzca la mortalidad por cáncer de mama. Asimismo, se ha demostrado que la RM combinada con la mamografía y el examen clínico de la mama tiene el más alto nivel de sensibilidad para detectar cáncer de mama en pacientes de alto riesgo.39
Cáncer contralateral de mama en portadoras BRCA1 y BRCA2
Después del diagnóstico inicial de cáncer de mama, el riesgo de cáncer contralateral es de aproximadamente 4% por año o 40% en 10 años.40 El riesgo es similar para las mutaciones BRCA1 o para las mutaciones BRCA2.41 Mientras más joven es la mujer en el momento en que se diagnostica el cáncer de mama, más alto es el riesgo de desarrollar en un futuro un cáncer contralateral.41 Debido al riesgo elevado, muchas portadoras tienden a optar por la mastectomía bilateral. La incidencia de cáncer contralateral se reduce con la ooforectomía y con la quimioterapia preventiva en aproximadamente 40%, a los 10 años.40,41 Es interesante destacar que ambos cánceres bilaterales, sincrónicos y asincrónicos, tienden a resemblar al otro en cuanto a grado y estatus del receptor estrogénico. La razón por la que esto ocurre aún permanece desconocida.1
Efectos de la terapia de reposición hormonal, anticonceptivos orales, paridad, lactancia y estilo de vida en las portadoras BRCA1 y BRCA2
Otros factores que pudiesen modificar el riesgo de desarrollar cáncer de mama o de ovario incluyen a los anticonceptivos orales, la terapia de reposición hormonal y el uso de drogas inductoras de la ovulación. Los anticonceptivos tienen un efecto protector para el cáncer de ovario, sin embargo, no tienen ningún efecto sobre el cáncer de mama.23,24,36 Por otro lado, la terapia de reposición hormonal y las drogas inductoras de la ovulación conllevan un riesgo elevado para cáncer de mama y de ovario, respectivamente.23,37
La paridad es un factor de riesgo para el cáncer de mama en portadoras del BRCA2, pero no en las portadoras del BRCA1.23,29
La lactancia materna por un periodo de un año o más (riesgo acumulativo) ha demostrado ser protectora en pacientes portadoras de ambos cánceres, mama y ovario.8, 29, 42 Por esta razón se debe promover la lactancia materna a las mujeres portadoras del BRCA1 y además se les debe aconsejar que lacten el mayor tiempo posible.
¿Cuál es el tratamiento más indicado una vez que el cáncer se ha desarrollado?
El tratamiento de las portadoras de mutaciones BRCA1 y BRCA2 afectadas con cáncer de mama difiere poco del de las pacientes con cáncer esporádico. Ahora bien, debido al elevado riesgo de desarrollar cáncer contralateral en las portadoras (39%)40 o de desarrollar cáncer de ovario (13%),30 algunas mujeres optan por la salpingo ooforectomía profiláctica o la mastectomía contralateral como parte de su tratamiento inicial.8
La radioterapia parece no incrementar el riesgo de cáncer en la mama contralateral,35 y la tasa de recurrencia ipsilateral después de la radioterapia es igual tanto en las portadoras como en las no portadoras.40 Los cánceres de mama asociados con BRCA, al igual que los no hereditarios, se benefician del uso de la radioterapia, pero el tejido mamario de las portadoras posee un riesgo indefinido debido a su predisposición hereditaria.8
Por lo antes expuesto, el tratamiento conservador es también una opción para las portadoras de mutaciones BRCA, pero debe seguirse de cerca por el riesgo que ellas tienen de desarrollar un segundo cáncer primario.8,21,32,33,35
Como es sabido, las mujeres portadoras de una mutación BRCA1 se asocian con cánceres de mama de alto grado, de tipo basal y con receptores estrogénicos negativos. Por lo tanto, estas pacientes son candidatas para tratamiento con quimioterapia.8 Estudios recientes sugieren que las portadoras del BRCA1 pudieran responder de una manera diferente a la quimioterapia neo-adyuvante o postoperatoria que las mujeres sin mutaciones.43 En Polonia, estudios realizados en portadoras de BRCA1 con cáncer de mama han encontrado que existe una respuesta a los taxanos mucho menor de la esperada, pero que, en cambio, estas pacientes responden mejor a tratamientos con cisplatino.43,44 La mayoría de los cánceres asociados con mutaciones en BRCA1 son receptores de estrógenos negativos, lo que implica que los tratamientos hormonales ablativos no están indicados en estas pacientes. Sin embargo, y como se mencionó anteriormente, la salpingo-ooforectomía es una buena medida preventiva tanto para el desarrollo de un cáncer primario como para las recurrencias locales o el cáncer contralateral.
Conclusión
El descubrimiento de los genes BRCA1 y BRCA2 en 1994 y 1995 ha llevado a la práctica la prevención oncológica y pruebas genéticas cada vez más sofisticadas para pesquisar pacientes de alto riesgo con cáncer de mama de origen hereditario. El beneficio de una prueba genética en cualquier población se debe a la habilidad de la misma de reducir tanto la incidencia como la mortalidad del cáncer de mama. La meta de la prueba genética es reducir el número de cánceres que aparezcan después de tener el resultado de la prueba, detectando los cánceres en un estadio temprano y ofreciendo óptimos tratamientos. Las mutaciones fundadoras permitirían agilizar la detección de portadoras y abaratar los costos. Es necesario unificar los criterios para saber a quién indicar las pruebas genéticas, crear pautas y recomendaciones y desarrollar políticas de salud pública.
En líneas generales, los genes que hasta la fecha han sido identificados como genes asociados con el cáncer hereditario de mama son BRCA1, BRCA2, Tp53, CHk2 y ATM. Todos ellos forman parta de la maquinaria celular que mantiene la integridad genómica y repara el ADN. Sin embargo, sólo las mutaciones en los genes BRCAson criterio para indicar una prueba genética.
El riesgo para desarrollar cáncer de mama es influenciado por la heterogeneidad de los alelos, por los genes modificadores y por cofactores ambientales y hormonales (anticonceptivos orales, terapia de reposición hormonal, paridad, lactancia.)
El seguimiento y vigilancia de las portadoras BRCAdebe incluir un autoexamen mensual de la mama desde los 25 años, examen clínico de la mama bianual y mamografías anuales o bianuales desde los 25-35 años; de ser posible se debe realizar la mamografía conjuntamente con la resonancia magnética, lo que aumentaría la sensibilidad y especificidad hasta en 95%. Dentro de las estrategias para la prevención del cáncer de mama se incluyen la quimioterapia profiláctica y la cirugía preventiva. La cirugía es una intervención en un solo tiempo y ofrece el mayor grado de protección. La ooforectomía está asociada con una reducción del riesgo de cáncer de ovario de 80% y con una reducción del riesgo de cáncer de mama hasta de 50%. Se recomienda ofrecer a toda mujer portadora de una mutación en BRCA1 o BRCA2 la salpingo-ooforectomía después de los 40 años de edad o una vez que haya cumplido su paridad, con el fin de reducir el riesgo. La mastectomía preventiva disminuye el riesgo de cáncer de mama en más de 90%. Si la cirugía está descartada por alguna razón, el tamoxifeno es una buena alternativa.
El mejor enfoque para el manejo de pacientes portadoras es el multidisciplinario, que incluya genetistas, consejeros genéticos, médicos oncólogos y cirujanos.
Declaración de conflicto de intereses: Los autores declararon no tener conflicto de intereses.
Referencias
1. Narod S, Foulkes W. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 2004; 4:665-676. [ Links ]
2. Volgestein B, Kinzler KW.The genetic basis of human cancer.2nd edition. McGraw Hill Co, 2002. [ Links ]
3.The Breast Cancer Linkage Consortium. Pathology of familial breast cancer: Differences between breast cancers in carriers of BRCA1 or BRCA2 mutations and sporadic cases. Lancet 1997;349: 1505-1510 [ Links ]
4. Antoniou A, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, Loman N, et al.Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies.Am J Hum Genet 2003; 72:1117-1130. [ Links ]
5. Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343:692-695. [ Links ]
6. Hall MJ, Reid JE, Burbidge LA, Pruss D, Deffenbaugh AM, Frye C, et al. Women of Different Ethnicities Undergoing Testing for Hereditary Breast-Ovarian Cancer. Cancer 2009;115(10):2222-2233. [ Links ]
7. Lynch H, Lynch J. Genética, Historia Natural y Consejo Genético basado en el ADN del Cáncer de Mama Hereditario. En:Winchester DJ, Winchester DP,American Cancer Society. Cáncer de mama.Atlas de Oncología Clínica. Ediciones Harcourt, 2001: 1-18. [ Links ]
8. Narod S, Offit K. Prevention and Management of Hereditary Breast Cancer. J Clin Oncol 2005; 23(8):1656-1663. [ Links ]
9.Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst 2006; 98:1694-1706. [ Links ]
10. Cazap E, Buzaid AC, Garbino C, de la Garza J, Orlandi FJ, Schwartsmann G, et al. Latin American and Caribbean Society of Medical Oncology. Breast cancer in Latin America: results of the Latin American and Caribbean Society of Medical Oncology/Breast Cancer Research Foundation expert survey. Cancer 2008 ;113(8 Suppl):2359-2365. [ Links ]
11. Metcalfe KA, Birenbaum-Carmeli D, Lubinski J, Gronwald J, Lynch H, Moller P. International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer 2008; 122:2017-2022. [ Links ]
12. Wooster R, Bignell G, Lancaster J. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378: 789-792 [ Links ]
13. John EM, Miron A, Gong G, Phipps AI, Felberg A, Li FP, et al. Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial groups. JAMA 2007; 298:2869-2876. [ Links ]
14. Malone K, Daling J, Doody D, Hsu L, Bernstein L, Coates R, et al. Prevalence and Predictors of BRCA1 and BRCA2 Mutations in a Population-Based Study of Breast Cancer in White and Black American Women Ages 35 to 64 Years. Cancer Res 2006; 66(16): 8297-8308. [ Links ]
15. Warner E, Foulkes W, Goodwin P, Meschino W, Blondal J, Paterson C, et al. Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst 1999;91:1241-1247. [ Links ]
16. Foulkes WD, Metcalfe K, Sun P, Hanna WM, Lynch HT, Ghadirian P, et al. Estrogen receptor status in BRCA1 and BRCA2 related breast cancer: the influence of age, grade, and histological type. Clin Cancer Res 2004:10:2029-2034. [ Links ]
17. Foulkes WD, Stefansson IM, Chappuis PO, Bégin LR, Goffin JR, Wong N, et al. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst 2003;95:1482-1485. [ Links ]
18.Lakhani SR, Reis-Filho JS, Fulford L, Penault-Llorca F, van der Vijver M, Parry S, et al. Breast Cancer Linkage Consortium. Prediction of BRCA1 status in patients with breast cancer using estrogens receptor and basal phenotype. Clin Cancer Res 2005; 11:5175-5180. [ Links ]
19.Young RS, Pilarski RT, Donenberg T, Shapiro S, Hammond LS, Miller J, et al.The Prevalence of BRCA1 Mutations Among Young Women with Triple-Negative Breast Cancer. BMC Cancer 2009; 9:86. [ Links ]
20. Dent R,Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al.Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Re 2o07,113:4429-4434. [ Links ]
21. Petrucelli N, Daly M, Culver J, Levy E. BRCA1 and BRCA2 Hereditary Breast/Ovarian Cancer. GeneReviews at Gene Tests: Medical Genetics Information Resource [database online], Seattle: University of Washington, 2005. [ Links ]
22. Morgan D, Sylvester H, Lucas FL, Miesfeldt S. Cancer prevention and screening practices among women at risk for hereditary breast and ovarian cancer after genetic counselling in the community setting. Familial Cancer 2009; 8:277-287. [ Links ]
23. Narod SA. Modifiers of risk of hereditary breast cancer. Oncogene 2006;25(43):5832-5836. [ Links ]
24. Finch A, Beiner M, Lubinski J, Lynch HT, Moller P, Rosen B. Salpingooophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 Mutation. JAMA 2006; 296:185-192. [ Links ]
25. Panchal S, Ennis M, Canon S. Bordeleau L. Selecting a BRCA risk assessment model for use in a familial cancer clinic. BMC Medical Genet 2008; 9:116. [ Links ]
26.Vogel KJ, Atchley DP, Erlichman J, Broglio KR, Ready KJ, Valero V, et al. BRCA1 and BRCA2 genetic testing in Hispanic patients: mutation prevalence and evaluation of the BRCAPRO risk assessment model. J Clin Oncol 2007;25:4635-4641. [ Links ]
27. Vidal-Millán S. Cáncer de Mama Hereditario: Identificación y Elección de Pacientes para Estudio Molecular de los Genes BRCA. Cancerología 2008;3:51-61. [ Links ]
28.The Am College of Obstet & Gynecol. Clinical Management guidelines for Obstetrician-Gynecologists 2009;113:4:957-966 [ Links ]
29. McLaughlin JR, Risch HA, Lubinski J, Moller P, Ghadirian P, Lynch H. Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet Oncol 2007;8:26-34. [ Links ]
30. Metcalfe KA, Lynch HT, Ghadirian P.The risk of ovarian cancer after breast cancer in BRCA1 and BRCA2 carriers. Gynecol Oncol 2005; 96:222-226. [ Links ]
31. Metcalfe KA, Finch A, Poll A, Horsman D, Kim-Sing C, Scott J, et al. Breast Cancer Risks in Women with a Family History of Breast or Ovarian Cancer who have Tested Negative for a BRCA1 or BRCA2. Mutation Br J Cancer 2008. [ Links ]
32. Eisen A, Rebbeck T,Wood W,Weberet B. Prophylactic Surgery in Women With a Hereditary Predisposition to Breast and Ovarian Cancer. J Clin Oncol 2000;18 (9): 1980. [ Links ]
33. Metcalfe KA, Lubinski J, Ghadirian P. Predictors of contralateral prophylactic mastectomy in women with a BRCA1 or BRCA2 mutation: the Hereditary Breast Cancer Clinical Study Group. J Clin Oncol 2008; 26:1093-1097. [ Links ]
34. Gronwald J, Tung N, Foulkes WD, Offit K, Gershoni R, Daly M. Tamoxifen and contralateral breast cancer in BRCA1 and BRCA2 carriers: an update. Int J Cancer 2006;118:2281-2284. [ Links ]
35. Gulati AP, Domchek SM.The Clinical Management of BRCA1 and BRCA2 Mutation Carriers. Curr Oncol Rep 2008;10(1):47-53. [ Links ]
36. Eisen A, Lubinski J, Klijn J, Moller P, Lynch HT, Offit K. Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: an international case-control study. J Clin Oncol 2005;23:7491-7496. [ Links ]
37. Eisen A, Lubinski J, Gronwald J, Moller P, Lynch HT, Klijn J. Hormone therapy and the risk of breast cancer in BRCA1 mutation carriers. J Natl Cancer Inst 2008;100:1361-1367. [ Links ]
38. Nelson H, Huffman L, Fu R, Harris E. Genetic Risk Assessment and BRCA Mutation Testing for Breast and Ovarian Cancer Susceptibility: Systematic Evidence Review for the U.S. Preventive Services Task Force. Ann Intern Med 2005; 143:362-379. [ Links ]
39. Warner E, Plewes DB, Hill KA, Causer PA, Zubovits JT, Jong RA. Surveillance of BRCA1 and BRCA2 mutation carriers with magnetic resonance imaging, ultrasound, mammography, and clinical breast examination. JAMA 2004;292:1317-1325. [ Links ]
40. Metcalfe K, Lynch HT, Ghadirian P,Tung N, Olivotto I,Warner E, et al. Contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2004;15:2328-2335. [ Links ]
41. Garber JE, Mehra G. Contralateral Breast Cancer in BRCA1/ BRCA2 Mutation Carriers:The Story of the Other Side. J Clin Oncol 2009;25.1652. [ Links ]
42. Jernström H, Lubinski J, Lynch HT, Ghadirian P, Neuhausen S, Isaacs. Breast-feeding and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 2004; 96:1094-1098. [ Links ]
43. Byrski T, Huzarski T, Dent R, Gronwald J, Zuziak D, Cybulski C, et al. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat 2008; 23. (epub ahead of print. [ Links ])
44. Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, et al. Polish Hereditary Breast Cancer Consortium. Response to neoadjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res Treat 2008;108:289-296. [ Links ]
45.Weitzel JN, Lagos V, Blazer KR, Nelson R, Ricker C, Herzog J, et al. Prevalence of BRCA mutations and founder effect in high-risk Hispanic families. Cancer Epi Biomarkers Prev 2005;14:1666-1671. [ Links ]
46.Weitzel JN, Lagos VI, Herzog JS, Judkins T, Hendrickson B, Ho JS, et al. Evidence for a common ancestral origin of a recurring BRCA1 genomic rearrangement identified in high-risk Hispanic families. Cancer Epi Biomarkers Prev 2007;16:1615-1620. [ Links ]
47. Gorski T, Byrski T, Huzarski TJ, Jakubowska A, Menkiszak J, Gronwald, et al. Founder mutations in the BRCA1 gene in Polish families with breastovarian cancer.Am J Hum Genet 2000;66:1963-1968. [ Links ]
48. Moller P, Borg A, Evans DG, Haites N, Reis MM,Vasen H, et al. Survival in prospectively ascertained familial breast cancer: analysis of a series stratified by tumor characteristics, BRCA mutations and oophorectomy. Int J Cancer 2002;101(6):555-559. [ Links ]
 Autor de correspondencia:
Autor de correspondencia:
Adriana A. Rodríguez
Women's College Research Institute
790 Bay Street,Toronto, ON, M5G 1NB, Canada
Correo electrónico: adriana.valentini@wchospital.ca
Fecha de aceptado: 27 de septiembre de 2011

