










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalud Pública de México
versión impresa ISSN 0036-3634
Salud pública Méx vol.52 no.5 Cuernavaca sep./oct. 2010
ARTÍCULO ORIGINAL
Receptores tipo Toll, patogénesis y respuesta inmune a Helicobacter pylori
Toll-like receptors, pathogenesis and immune response to Helicobacter pylori.
Norma Angélica Sánchez-Zauco, QBP(I); Silvia Giono-Cerezo, D en C(I); Carmen Maldonado-Bernal, D en C.(II)
(I) Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional. México, DF, México.
(II) Unidad de Investigación Médica en Enfermedades Oncológicas, Hospital Infantil de México Federico Gómez. México, DF, México.
Resumen
Helicobacter pylori coloniza el epitelio gástrico y la mayoría de las personas infectadas es asintomática, de 10 al 20% desarrolla gastritis atrófica, úlcera péptica, y menos de 3% genera cáncer gástrico. Estas patologías están determinadas por la relación entre los factores de virulencia de la bacteria y los factores del hospedero como predisposición genética y respuesta inmune. La inmunidad innata, representada principalmente por los receptores tipo Toll y tipo Nod, reconocen a sus ligandos específicos y activan factores de transcripción como NF-kB, AP-1, CREB-1, induciendo la producción de citocinas inflamatorias como IL-8, IL-12, IL-6, IL-1β, IL-18 y TNF-α, e IL-10. La inflamación crónica favorece los cambios de morfología gástrica, evita la apoptosis y favorece la angiogénesis, ocasionando lesiones neoplásicas y cáncer. El objetivo de esta revisión es analizar los mecanismos propuestos a la fecha de la respuesta inmune innata y adaptativa, involucrados en la infección por H. pylori, y se puntualiza en los mecanismos de eliminación o persistencia de la infección.
Palabras clave: inmunidad innata; inmunidad adaptativa; Helicobacter pylori /patogenecidad
Abstract
Helicobacter pylori colonize the gastric epithelial, most infected people are asymptomatic, 10 to 20% develop atrophic gastritis, peptic ulcer and less than 3% gastric cancer. These diseases are determined by the relationship between virulence factors of bacteria, host factors such as, genetic predisposition, and immune response. The innate immune response mainly represented by Toll-like receptors and Nod-like receptors that recognize their specific ligands, activate transcription factors as NF-kB, AP-1, CREB-1, inducing production of inflammatory cytokines such as IL -8, IL-12, IL-6, IL-1β, IL-18, TNF-α and IL-10. Chronic inflammation promotes gastric morphological changes, prevents apoptosis and allows angiogenesis generating neoplasic lesions and cancer. The aim of this review is to analyze the mechanisms proposed to date of the innate and adaptative immune response involved in H. pylori infection; remarking the mechanisms related in the elimination or persistence.
Key words: immunity, inate; adaptative immunity; Helicobacter pylori /pathogenicity
Helicobacter pylori es una bacteria microaerofílica, gram-negativa y flagelar que coloniza la mucosa gástrica. En países desarrollados infecta de 25 a 50% de la población y en países en vía de desarrollo de 70 a 90%.1 La mayoría de los individuos infectados son asintomáticos, pero de 10 a 20% pueden desarrollar gastritis atrófica, úlcera péptica, adenocarcinoma gástrico o linfoma asociado a mucosa (MALT), y <3% cáncer gástrico.2 H. pylori ha sido clasificada como un carcinógeno de tipo I.3
El desarrollo de las diferentes patologías depende de la predisposición genética del hospedero y de su respuesta inmune. Además, participan factores de virulencia de la bacteria y del medio ambiente como dieta y estilo de vida.4
Dentro de los factores de virulencia de H. pylori se encuentra la isla de patogenicidad cag (cag PAI) de 40 kb, que contiene un sistema de secreción tipo IV (T4SS); que transloca CagA, proteína efectora, oncogénica. La cag PAI está asociada con daño al tejido y riesgo de padecer enfermedades graves como úlcera, cáncer gástrico y MALT.4
Inmunidad innata
El sistema inmune innato censa, responde y modula la respuesta a microorganismos y a productos microbianos por medio de receptores que están codificados en la línea germinal, entre ellos los receptores tipo Toll (TLR) y tipo NOD (NLR). Los TLR son una familia de moléculas de superficie y citoplasmáticas que participan en el reconocimiento de patrones moleculares asociados a microorganismos (MAMP) y patrones moleculares asociados a daño (DAMP). Originalmente se describió a la proteína Toll como la responsable de la polaridad dorsoventral en el embrión de Drosophila y en la mosca adulta, participando en la protección de infecciones fúngicas.5 En mamíferos se han descrito 13 receptores homólogos a Toll (TLR).5 Los TLR son receptores transmembranales tipo I; tienen un dominio extracelular con regiones repetitivas ricas en leucina (LRR), de 24 a 29 aminoácidos; tienen una o dos regiones ricas en cisteína, además de un dominio intracelular de aproximadamente 200 aminoácidos, similar al descrito en la familia de receptores de la interleucina1 (IL-1R), denominado Toll/IL-1R (TIR), por medio del cual se lleva a cabo la transducción de señales (figura 1).6
TLR2 reconoce peptidoglicanos (PGN), ácido lipoteicoico (LTA) y lipoproteínas de bacterias Gram positivas, así como -œzimosan- de levaduras. TLR3 reconoce RNA de doble cadena (dsRNA). TLR4 reconoce el lipopolisacárido (LPS) de la pared de bacterias Gram negativas, las proteínas de choque térmico (HSP) de 60 y 70 kDa, entre otros. TLR5 reconoce flagelina bacteriana y TLR9 participa en el reconocimiento de CpGs, de DNA no metilado. TLR1, TLR6 y TLR10 pueden formar heterodímeros con TLR2 aumentando la especificidad por sus ligandos.7
Los TLR representan un puente entre el sistema inmune innato y el sistema inmune adaptativo.6 Ya que la activación del sistema inmune innato induce la fagocitosis, la opsonización y la producción de mediadores de la inflamación, bloqueando la diseminación del patógeno. Los TLR y NLR que están expresados en las células presentadoras de antígeno (APC), tras reconocer a sus ligandos, se activan e inducen moléculas que participan en la presentación de péptidos antigénicos sobre su superficie. En el complejo principal de histocompatibilidad (MHC) los péptidos son reconocidos por las células T antígeno específicas, uniéndose así la respuesta inmune innata y la adaptativa (figura 1).6,8
El reconocimiento de MAMP y DAMP intracelulares también puede ser por una familia de receptores citoplásmicos denominados potenciadores de la transcripción, Nod-repetidos ricos en leucina, proteínas CATERPILLER o dominios de oligomerización a nucleótidos (NOD).9
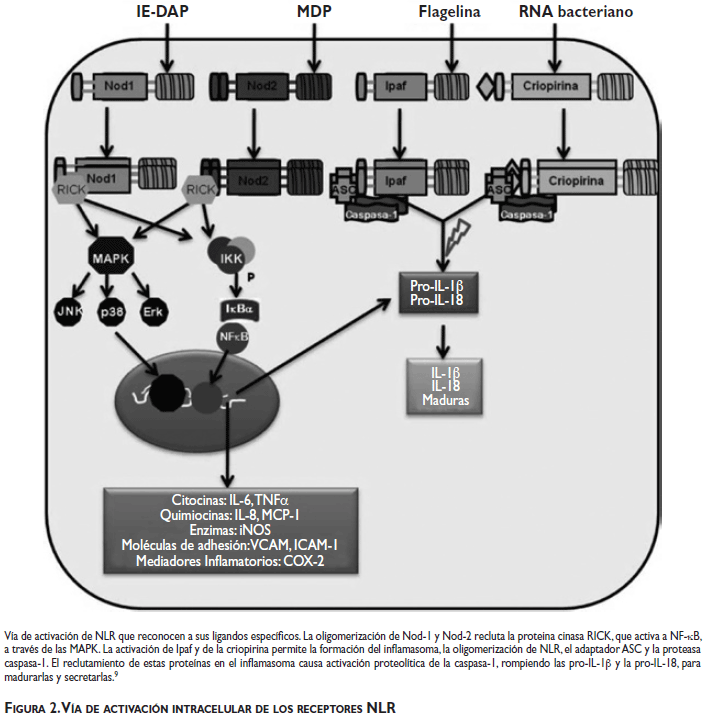
Nod-1 y Nod-2 inducen la expresión y la secreción de citocinas proinflamatorias, incluyendo al factor de necrosis tumoral alfa (TNF-α) (figura 2).9
En respuesta a H. pylori se activa NF-kB dependiente de Nod-1, induciéndose β-defensinas y quimiocinas como CXCL1, CXCL2 y CCL2 que reclutan neutrófilos -œin vivo-. Los ratones deficientes en Nod-1 infectados con H. pylori se colonizan con un mayor número de bacterias, comparados con ratones silvestres, por la respuesta débil de las quimiocinas (figura 2).10
Las células epiteliales gástricas tienen un mecanismo intracelular para detectar a bacterias no invasivas. H. pylori inyecta muropéptidos dentro de las células mediante el sistema de secreción tipo IV (T4SS). La liberación de ácido diaminopimélico g-D-glutamil (iE-DAP) es suficiente para la activación de Nod-1; sin embargo, también se inyectan moléculas de PGN que activan a Nod-2, el cual contribuye a la síntesis y secreción de α-defensinas (figura 2).9
H. pylori y la generación de la respuesta inmune
La inducción de mediadores proinflamatorios, después del contacto con los TLR que se encuentran en la superficie de las células epiteliales, indica que no sólo son una barrera física, sino que son fundamentales para activar al sistema inmune innato por el reconocimiento de MAMP.11
El LPS de H. pylori estimula al TLR4, induciendo la expresión del gen de la mitógeno oxidasa-1 en células gástricas de cobayo.12 Sin embargo, se ha observado que el cultivo celular primario de biopsias de antro no expresan TLR4, o lo expresan muy débilmente, lo que indica que otras células presentes en la mucosa pudieran ser las que reconocen a H. pylori.11 El TLR4 de diferentes especies presenta diferencias en el reconocimiento del LPS. El TLR4 de ratones y cobayos, pero no de humanos, reconoce LPS tetra y penta-acilados (hipoacilados), lo cual explica por qué H. pylori causa infecciones crónicas en humanos pero no infecta a ratones.13 El lípido A del LPS de H. pylori es una mezcla de estructuras tetra-aciladas y hexa-aciladas.14 El LPS de H. pylori es similar al de P. gingivalis, que es reconocido por TLR2. Ambos LPS se unen débilmente a la proteína unidora de LPS (LBP) y se transfieren débilmente a CD14.15
El LPS de H. pylori que expresa la forma monomérica del antígeno Le se une al dominio de reconocimiento de carbohidratos DC-SIGN, el cual está en la superficie de células dendríticas presentes en la mucosa gástrica; esta interacción afecta la polarización de la respuesta celular Th1/Th2.16
Las células epiteliales de antro y de cuerpo del estómago humano expresan TLR2, TLR4, TLR5 y TLR9, los cuales se encuentran en el polo basolateral y apical, mientras que en pacientes infectados con H. pylori los TLR 4, 5 y 9 se concentran en el polo apical.17
Los estudios sugieren que el TLR2, más que TLR4, participa en el reconocimiento de H. pylori y que la expresión del TLR4 y MD-2 se modifica en pacientes infectados con H. pylori.1 El reconocimiento del LPS necesita del correceptor CD14, que está en dos formas: una anclada a la membrana (CD14m), presente en la superficie de monocitos/macrófagos y neutrófilos, y otra en forma soluble (CD14s), que se produce en hígado y está en suero y orina. CD14s transporta el LPS a la proteína unidora de LPS (LPB) y no al complejo TLR4, MD2, CD14m, con disminución de respuesta Th1. El CD14 se considera modulador de apoptosis inducida por el LPS en células epiteliales y endoteliales.18 El polimorfismo en el promotor de CD14 afecta su expresión y estimula la persistencia de la infección de H. pylori, lo que aumenta el riesgo de cáncer gástrico.19
La proteína de choque térmico (HSP60) de H. pylori activa a TLR2 y TLR4, incrementando la actividad de NF-kB e induciendo IL-8 en células epiteliales gástricas.20
El T4SS codificado por cag PAI inyecta flagelina monomérica del citosol de la bacteria al citosol de las células eucariotas, participando en el progreso de la enfermedad.21 El TLR5 reconoce dominios conservados de flagelina, induciendo la expresión de citocinas proinflamatorias e IL-8. La flagelina en el citosol de macrófagos se detecta por Ipaf, que activa a la caspasa 1 para producir IL-1β e IL-18. La inducción de IL-1β requiere reconocimiento del ligando por el TLR5, activando el NLR para inducir su secreción.21
La estimulación de TLR2 altera la expresión del TLR5 y, al estimular al TLR4 con LPS, aumenta la expresión del TLR2.22
A través del T4SS, H. pylori libera DNA, el cual es reconocido por TLR9 que se expresa en la superficie de las células gástricas.17 El heterodímero TLR2/TLR9 induce la activación de la vía NF-kB/PKC/c-Src, que favorece la expresión de COX-2, que activa a las MAPK como ERK1/2, p38, JNK.23 éstas activan a los factores de transcripción CREB-1, ATF-2, c-fos, y c-jun, que activan a AP-1 y CRE sobre el promotor de COX-2.23 Estas cascadas inducen la producción de IL-8 y liberación de PGE2 dependiente de COX-2. Además, alteran la expresión de genes como la metaloproteasa de matriz-7, que degrada la matriz extracelular y procesa sustratos bioactivos como FAS-L, integrina β4, E-cadherina, pro-EGF y plasminógeno, necesarios en la epitelización, apoptosis, inflamación y respuesta inmune innata.23
La expresión aumentada de COX-2 está implicada en diferentes tipos de cáncer, como el pulmonar, esofágico, pancreático, de cerebro, colorrectal y gástrico, debido a que es un factor que protege a las células de apoptosis, permite la proliferación celular, invasión de células tumorales y angiogénesis e inhibe la vigilancia inmunológica.23,24 Pacientes con cáncer gástrico y úlcera péptica infectados con H. pylori presentan elevada expresión de COX-2 y PGE2. El empleo de anticuerpos anti-TLR2 y anti-TLR9 impide la expresión de COX-2 inducida por H. pylori en células epiteliales gástricas humanas.23
La respuesta proinflamatoria inducida por H. pylori resulta en la activación de factores de transcripción como NF-kB y AP-1. A su vez, modifica la expresión de diversas citocinas proinflamatorias y quimiocinas, entre ellas IL-8. Además, la activación de NF-kB mejora la expresión y exposición de la molécula de adhesión intercelular 1 (ICAM-1), la cual está presente en la superficie del endotelio y de los leucocitos. ICAM-1, cuando se une a su receptor, el antígeno asociado a la función del leucocito 1 (LFA-1), produce la agregación homotípica de las células TPH1 (células monocíticas humanas) infectadas con H. pylori.25
El polimorfismo de TLR se ha asociado con un aumento en la susceptibilidad o severidad de las infecciones.26,27 El polimorfismo del TLR4 en Asp299Gly y Thr399Ile en pacientes infectados con H. pylori es un factor de riesgo de cáncer gástrico tipo intestinal en población italiana,28 y en población mexicana fue más frecuente en úlcera duodenal y cáncer gástrico, comparado con casos de gastritis superficial.27 El cambio en el alelo 896 de TLR9 está asociado con gastritis y metaplasia en población Hindú29 y el polimorfismo en el promotor del gen de TLR9 (TLR9-123T/C) crea un sitio de unión al NF-kB, incrementando la actividad transcripcional, lo que se ha asociado con la inducción de cambios gástricos premalignos inducidos por H. pylori.30
Patogénesis de Helicobacter pylori
En respuesta a H. pylori aumenta el RNAm de numerosas citocinas como TNFα, IL-1β, IL-6, IL-8, IL-10, IFN-γ e IL-18.31,32 La producción de IL-8, relacionada con la respuesta inmune innata y adaptativa frente a H. pylori, aumenta la permeabilidad celular, recluta y activa neutrófilos y aumenta la interacción de la bacteria con los macrófagos y células pertenecientes al linaje linfoide.33 De la misma manera, la IL-6 se incrementa en respuesta a la bacteria e induce inflamación crónica, con infiltración de PMN y células mononucleares (MNC).32,33 La unión de H. pylori con antígeno de Lewis en el LPS al DC-SIGN reduce la cantidad de IL-6.16
H. pylori aumenta la producción de TNF-α, con producción de IL-8, debido a una regulación positiva que existe sobre TNF-α e IL-1β.34 TNF-α contribuye al aumento de la apoptosis, vía caspasa-3, con activación de caspasa-8.34 IL-12 se produce por macrófagos y monocitos, después de la estimulación con antígenos bacterianos, y da inicio a la cascada inflamatoria, lo que aumenta el grado de gastritis.32,35 H. pylori también activa a células NK, que producen IFN-γ, aumentando la expresión de moléculas del complejo principal de histocompatibilidad tipo II, e induce apoptosis.34 De la misma forma, la inflamación gástrica disminuye y regula la colonización y activa macrófagos; éstos secretan citocinas proinflamatorias y modifican la expresión de citocinas antiinflamatorias como TGF-β.31
Las citocinas modulan la expresión de hormonas gástricas que controlan la secreción de ácido gástrico. El IFN-γ incrementa la gastrina, estimulando la secreción de ácido gástrico, mientras que la IL-4 aumenta la producción de somatostatina, en células D, disminuyendo la secreción de gastrina, con una reducción severa de la gastritis.36
La mucosa gástrica inflamada presenta centros germinales con agregados linfoides; en niños infectados con H. pylori se genera gastritis crónica que se denomina gastritis folicular.37 Las células B, activadas por la linfopoyetina del estroma del timo (TSLP), por MIP-3a (quimiocina que atrae a células dendríticas) y por el factor activador de células B (BAFF), favorecen la producción de moléculas coestimuladoras CD80, que al unirse a su receptor, CD28 (células TCD4+), produce citocinas Th2 como IL-4 e IL-13 y citocinas pro-inflamatorias TNFα e IFN-γ.37 Las células plasmáticas producen anticuerpos, especialmente de clase IgA secretora, la cual no activa al complemento y por la adherencia a la bacteria permite que evada la respuesta inmune. En humanos, la IgA es de la subclase IgA1, que no es polimérica y carece de la cadena J, por lo que es inestable y susceptible de degradación por proteasas; esto y los bajos niveles de IgA e IgG que se unen a la superficie de la bacteria, evitan la activación de la respuesta inmune. La unión de los anticuerpos al receptor Fcγ en los leucocitos tiene una función inhibitoria y promueve la liberación de citocinas antiinflamatorias como IL-10.38
Los pacientes infectados crónicos presentan en el suero y en la mucosa gástrica títulos elevados de anticuerpos específicos de clase IgG e IgA. Sin embargo quedan infectados, persistiendo H. pylori.38
El polimorfismo de IL-1β y de su receptor (IL-1RN) incrementan el riesgo de adenocarcinoma de cardias en personas infectadas con H. pylori. El polimorfismo en la región promotora de TNF-α y el solo cambio de un nucleótido 308G>A también incrementan el riesgo de cáncer gástrico.39 La IL-16 es una citocina producida por los monocitos que promueve la secreción de citocinas inflamatorias asociadas a tumores. Un cambio en rs11556218 T/G se ha asociado con mayor susceptibilidad a cáncer gástrico.40 El IFN-γ promueve la gastritis. Individuos homocigotos para el alelo IFNGR1-56*T presentan cuatro veces mayor riesgo de padecer cáncer gástrico, en comparación con el homocigoto para el alelo IFNGR1-56*C.39
Las biopsias de antro gástrico presentan infiltrado de linfocitos y monocitos que incrementan la expresión de IL-1β, IL-8 e IL-6.41 A las cuatro semanas aumenta el número de células T CD4+ y CD8+; sin embargo, desde las 42-72h hay expansión clonal de células T CD4+, lo que indica una respuesta inmune adaptativa temprana.41 En la mayoría de las personas se desarrolla -œgastritis superficial crónica-, en la que se presentan mayor cantidad de linfocitos CD4+ que de CD8+ y leucocitos polimorfonucleares (PMN). La infiltración de PMN es importante en la patogénesis, por citotoxicidad directa, al liberar agentes oxidativos y elastasa.42 La presencia de células T reguladoras (CD4+/CD25+/Foxp3+) indica que hay regulación de la respuesta inmune. En la inflamación crónica hay un efecto combinado de una respuesta inmune celular y una estimulación constante de la respuesta inmune innata.43 Además, se reconocen epitopos de ATPasa por las células T, lo que contribuye al desarrollo de gastritis autoinmune.44
Inmunoprofilaxis
Actualmente los inmunoprofilácticos están elaborados a partir de antígenos involucrados en la patogénesis de la infección, como VacA, CagA, HP-NAP, ureasa, HP0231, adhesinas (BabA y SabA), o bien son lisados bacterianos unidos a la toxina colérica como adyuvante y se han probado en animales, previniendo la infección experimental. Sólo algunas vacunas se han probado en humanos, sin embargo no confieren protección adecuada. También se han empleado adyuvantes como el hidróxido de aluminio, con lo cual se genera una vacuna prometedora.45 Esta última formulación induce la respuesta de células T con la producción de IFN-γ, MIP-2, CXCL5, y CXCL1, las cuales atraen al estómago a los neutrófilos, células importantes en la erradicación de H. pylori. La respuesta de anticuerpos específicos no es suficiente para la protección contra H. pylori; también se requiere la respuesta celular.46 Recientemente se han usado oligonucleótidos CpG sonicados como vacunas, los cuales inducen una fuerte respuesta sistémica y local de tipo Th1.47
Conclusiones
H. pylori coloniza y persiste en el estómago humano y genera patologías severas como el cáncer y la úlcera péptica, a pesar del desarrollo de una fuerte respuesta inmune innata, celular y humoral, que involucra la participación de los TLR y NLR.
Sabemos que existe comunicación entre la bacteria y las células de la respuesta inmune, la cual es necesario entender para plantear alternativas terapéuticas o desarrollar vacunas para proteger de la infección o frenar el daño que ocasiona la respuesta inflamatoria asociada a la bacteria. Comprender esa comunicación permitirá pronosticar la evolución de la enfermedad y tomar las medidas adecuadas de forma oportuna.
Declaración de conflicto de intereses
Declaramos no tener conflicto de intereses.
Referencias
1. Smith MF, Mitchell A, Li G, Ding S, Fitzmaurice MA, Ryan K, et al. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required of Helicobacter pylori-induced NF-kB activation and chemokine expression by epithelial cells. J Biol Chem 2003;278(35):32552-32560. [ Links ]
2. Correa P, Piazuelo MB, Camargo MC. Etiopathogenesis of gastric cancer. Scand J Surg 2006;95(4):218-224. [ Links ]
3. International Agency for Research on Cancer: Schitosomes, liver flukes and Helicobacter pylori. Monographs on the evaluation of carcinogenic risks to humans Lyon: IARC, 1994:61. [ Links ]
4. Dorer MS, Talarico S, Salama NR. Helicobacter pylori's unconventional role in health and disease. PLoS Pathog 2009;5(10):1-6. [ Links ]
5. Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science 2002;296(5566):298-300. [ Links ]
6. Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature 2000;406(6797):782-787. [ Links ]
7. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124(4):783-801. [ Links ]
8. Takeda K, Kaisho T, Akira S. Toll like receptors. Annu Rev Immunol 2003; 21:335-376. [ Links ]
9. Franchi L, McDonald C, Kanneganti TD, Amer A, Núñez G. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol 2006;177:3507-3513. [ Links ]
10. Sanderson IR, Walker WA. TLRs in the Gut I. The role of TLRs/Nods in intestinal development and homeostasis. Am J Physiol Gastrointest Liver Physiol 2007;292:G6(1)-G10. [ Links ]
11. Bäckhed F, Rokbi B, Torstensson E, Zhao Y, Nilsson C, Seguin D, et al. Gastric mucosal recognition of Helicobacter pylori is independent of Toll-Like receptor 4. J Infect Dis 2003;187(5):829-836. [ Links ]
12. Kawahara T, Teshima S, Oka A, Sugiyama T, Kishi K, Rokutan K. Type I Helicobacter pylori lipopolysaccharide stimulates Toll-Like receptor 4 and activates mitogen oxidase-1 in gastric pit cells. Infect Immun 2001;69:4382-4389. [ Links ]
13. Hajjar Am, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human Toll-like receptor 4 recognizes host-specific LPS modification. Nat Immunol 2002;3:354-359. [ Links ]
14. Moran AP, Lindner B, Walsh EJ. Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol 1997;179:6453-6463. [ Links ]
15. Cunningham MD, Seachord C, Ratcliffe K, Bainbridge B, Aruffo A, Darveau RP. Helicobacter pylori and Porphyromonas gingivalis lipopolysaccharides are poorly transferred to recombinant soluble CD14. Infect Immun 1996;64(9):3601-3608. [ Links ]
16. Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth HP, et al. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med 2004;200(8):979-990. [ Links ]
17. Schmausser B, Andrulis M, Endrich S, Lee SK, Josenhans C, Muller-Hermelink HK, et al. Expression and subcellular distribution of toll-like receptors TLR4, TLR5 and TLR9 on the gastric epithelium in Helicobacter pylori infection. Clin Exp Immunol 2004;136(3):521-526. [ Links ]
18. Pugin J, Schür-Maly CC, Leturcq D, Moriarty A, Ulevitch RJ, Tobias PS. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc Natl Acad Sci USA 1993;90(7):2744-2748. [ Links ]
19. Zhao D, Sun T, Zhang X, Guo Y, Yu D, Yang M, et al. Role of CD14 promoter polymorphisms in Helicobacter pylori infection related gastric carcinoma. Clin Cancer Res 2007;13(8):2362-2368. [ Links ]
20. Zhao Y, Yokota K, Ayada K, Yamamoto Y, Okada T, Shen L, et al. Helicobacter pylori heat-shock protein 60 induces interleukin-8 via a Toll-like receptor (TLR)2 and mitogen-activated protein (MAP) kinase pathway in human monocytes. J Med Microbi 2007;56(Pt2):154-164. [ Links ]
21. Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, et al. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci USA 2005;102(26):9247-9252. [ Links ]
22. Cabral ES, Gelderblom H, Hornung RL, Munson PJ, Martin R, Marques AR. Borrelia burgdorferi lipoprotein-mediated TLR2 stimulation causes the down-regulation of TLR5 in human monocytes. J Infect Dis 2006;193(6):849-859. [ Links ]
23. Chang YJ, Wu MS, Lin JT, Chen CC. Helicobacter pylori-induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J Immunol 2005;175(12):8242-8252. [ Links ]
24. Huang M, Stolina M, Sharma S, Mao JT, Zhu L, Miller PW, et al. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res 1998;58(6):1208-1216. [ Links ]
25. Bauer B, Moese S, Bartfeld S, Meyer TF, Selbach M. Analysis of cell type-speciïc responses mediated by the type IV secretion system of Helicobacter pylori. Infect Immun 2005;73(8):4643-4652. [ Links ]
26. Moura SB, Almeida LR, Guerra JB, Rocha GA, Camargos-Rocha AM, Melo FF, et al. Toll-like receptor (TLR2, TLR4 and TLR5) gene polymorphisms and Helicobacter pylori infection in children with and without duodenal ulcer. Microbes In Infect 2008;10(14-15):1477-1483. [ Links ]
27. Trejo-de la O A, Torres J, Pérez-Rodríguez M, Camorlinga-Ponce M, Luna LF, Abdo-Francis JM, et al. TLR4 single-nucleotide polymorphisms alter mucosal cytokine and chemokine patterns in Mexican patients with Helicobacter pylori-associated gastroduodenal diseases. Clin Immunol 2008;129(2):333-340. [ Links ]
28. Santini D, Angeletti S, Ruzzo A, Dicuonzo G, Galluzzo S, Vincenzi B, et al. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in gastric cancer of intestinal and diffuse histotypes. Clin Exp Immunol 2008;154(3):360-364. [ Links ]
29. Achyut BR, Ghoshal UC, Moorchung N, Mittal B. Association of Toll-like receptor-4 (Asp299Gly and Thr399Ileu) gene polymorphisms with gastritis and precancerous lesions. Hum Immunol 2007;68(11):901-907. [ Links ]
30. Ng Mt, Van't Hof R, Crockett JC, Hope ME, Berry S, Thomson J, et al. Increase in NF-{kappa}B binding affinity of the C allelic variant of the Toll-like receptor 9-1237T/C polymorphism is associated with Helicobacter pylori induced gastric disease. Infect Immun 2010;78:1345-1352. DOI:10.1128/IAI.01226-09 [ Links ]
31. Straubinger RK, Greiter A, McDonough PS, Gerold A, Scanziani E, Soldati S, et al. Quantitative evaluation of inflammatory and immune responses in the early stages of chronic Helicobacter pylori infection. Infect Immun 2003;71(5):2693-2703. [ Links ]
32. Karttunen RA, Karttunen TJ, Yousfi MM, el-Zimaity HM, Graham DY, el-Zaatari FA. Expression of mRNA for interferon-gamma, interleukin-10, and interleukin-12 (p40) in normal gastric mucosa and mucosa infected with Helicobacter pylori. Scand J Gastroenterol 1997;32(1):22-27. [ Links ]
33. Agnihotri N, Bhasin DK, Vohra H, Ray P, Singh K, Ganguly NK. Characterization of lymphocytic subset and cytokine production gastric biopsy samples from Helicobacter pylori patients. Scand J Gastroenterol 1998;33(7):704-709. [ Links ]
34. Noach LA, Bosma NB, Jansen J, Hock FJ, van Deventer SJ, Tytgat GN. Mucosal tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 production in patients with Helicobacter pylori infection. Scand J Gastroenterol 1994;29(5):425-429. [ Links ]
35. Bauditz J, Ortner M, Bierbaum M, Niedobitek G, Lochs H, Schreiber S. Production of IL-12 in gastritis relates infection with Helicobacter pylori. Clin Exp Immunol 1999;117(2):316-323. [ Links ]
36. Zavros Y, Rathinavelu S, Kao JY, Todisco A, Del Valle J, Weinstock JV, et al. Treatment of Helicobacter gastritis with IL-4 requires somatostatin. Proc Natl Acad Sc USA 2003;100(22):12944-12949. [ Links ]
37. Kido M, Tanaka J, Aoki N, Iwamoto S, Nishiura H, Chiba T, et al. Helicobacter pylori promotes the production of thymic stromal lymphopoietin by gastric epithelial cells and induces dendritic cell-mediated inflammatory Th2 responses. Infect Immun 2010;78(1):108-114. [ Links ]
38. Akhiani AA., Schön K, Franzén LE, Pappo J, Lycke N. Helicobacter pylori-speciïc antibodies impair the development of gastritis, facilitate bacterial colonization, and counteract resistance against infection. J Immunol 2004;172(8):5024-5033. [ Links ]
39. Correia M, Machado JC, Ristimaki A. Basic aspects of gastric cancer. Helicobacter 2009;14 Suppl1:36-40. [ Links ]
40. Gao LB, Rao L, Wang YY, Liang WB, Li C, Xue H, et al. The association of interleukin-16 polymorphisms with IL-16 serum levels and risk of colorectal and gastric cancer. Carcinogenesis 2009;30(2):295-299. [ Links ]
41. Nurgalieva ZZ, Conner ME, Opekun AR, Zheng CQ, Elliott SN, Ernst PB, et al. B-cell and T-cell immune responses to experimental Helicobacter pylori infection in humans. Infect Immun 2005;73(5):2999-3006. [ Links ]
42. Hofman V, Ricci V, Galmiche A, Brest P, Auberger P, Rossi B, et al. Effect of Helicobacter pylori on polymorphonuclear leukocyte migration across polarized T84 epithelial cell monolayers: role of vacuolating toxin VacA and cag pathogenicity island. Infect Immun 2000;68(9):5225-5233. [ Links ]
43. Peek RM. Events at the host-microbial interface of the gastrointestinal tract IV. The pathogenesis of Helicobacter pylori persistence. AM J Physiol Gastrointest Liver Physiol 2005;289:G8-G12. [ Links ]
44. D'Elios MM, Bergman MP, Amedei A, Appelmelk BJ, Del Prete G. Helicobacter pylori and gastric autoimmunity. Microbes Infect 2004;6(15):1395-1401. [ Links ]
45. Aebischer T, Bumann D, Epple HJ, Metzger W, Schneider T, Cherepnev G, et al. Correlation of T cell response and bacterial clearance in human volunteers challenged with Helicobacter pylori revealed by randomised controlled vaccination with Ty21a-based Salmonella vaccines. Gut 2008;57:1065-1072. [ Links ]
46. Del Giudice G, Covacci A, Telford J, Montecucco C, Rappuoli R. The design of vaccines against Helicobacter pylori and their development. Annu Rev Immunol 2001;19:523-563. [ Links ]
47. Taylor JM, Ziman ME, Canfield DR, Vajdy M, Solnick JV. Effects of a Th1- versus a Th2-biased immune response in protection against Helicobacter pylori challenge in mice. Microb Pathog 2008;44:20-27. [ Links ]
Solicitud de sobretiros: Dra. Silvia Giono Cerezo. Laboratorio de Bacteriología Médica, Escuela Nacional de Ciencias Biológicas,
Instituto Politécnico Nacional. Plan de Ayala y Carpio S/N, Col. SantoTomás. 11340, Delegación Miguel Hidalgo. México, DF.
Correo electrónico: sgiono@yahoo.com
Fecha de recibido: 14 de enero de 2010 • Fecha de aceptado: 24 de junio de 2010


{kind=link}
{kind=link}