










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalud Pública de México
versión impresa ISSN 0036-3634
Salud pública Méx vol.51 supl.2 Cuernavaca ene. 2009
ARTÍCULO DE REVISIÓN
Bases genómicas del cáncer de mama: avances hacia la medicina personalizada
Genomic basis for breast cancer: advances in personalized medicine
Alfredo Hidalgo-Miranda, PhD; Gerardo Jiménez-Sánchez, MD, PhD
Instituto Nacional de Medicina Genómica. México
RESUMEN
El análisis genómico del cáncer de mama ha permitido el desarrollo de nuevas herramientas de predicción de riesgo y respuesta al tratamiento en esta enfermedad. Los perfiles de expresión génica han generado una mejor clasificación de los tumores e identificado subgrupos tumorales con características clínicas particulares. También se han reconocido patrones de pérdida y ganancia de DNA y expresión de micro-RNA relacionados con la carcinogénesis mamaria, tras identificar nuevos blancos potenciales. Los estudios de asociación del genoma completo han identificado variantes genéticas vinculadas con un mayor riesgo a presentar esta enfermedad, lo que hará posible tomar decisiones de salud pública mejor fundamentadas. Asimismo, los avances en la tecnología de secuenciación de DNA permitirán obtener información acerca de todas las alteraciones genéticas en los tumores. En esta revisión se describe el estado que guarda la investigación genómica en el cáncer de mama, así como la transición de estos hallazgos a la práctica clínica y la creación de las bases para el desarrollo de la medicina personalizada.
Palabras clave: cáncer; mama; medicina genómica; expresión; microarreglos
ABSTRACT
Genomic analysis of breast cancer has allowed the development of new tools for the prediction of recurrence and the response to treatment of this disease. Gene expression profiles allow better tumor classification, identifying tumor subgroups with particular clinical outcomes. New potential molecular targets involved in breast carcinogenesis have also been identified through the analysis of DNA copy number aberrations and microRNA expression patterns. Whole genome association studies have identified genetic variants associated with a higher risk to develop this tumor, providing more information for public health decisions. Progress in DNA sequencing methods will also allow for the analysis of all the genetic alterations present in a tumor. In this review, we describe the current state of genomic research in breast cancer as well as how these findings are being translated into clinical practice, contributing to development of personalized medicine.
Key words: cancer; breast; genomic medicine; expression; microarrays
En México, el cáncer de mama es a partir del año 2006 la segunda causa de muerte en mujeres de 30 a 54 años.1 Sólo en este año 4 451 mujeres murieron por esta neoplasia, se reconocieron casos de cáncer de mama desde la segunda década de vida y se registró una incidencia máxima entre los 40 y 54 años. En promedio, las mujeres mexicanas desarrollan cáncer de mama una década antes que las europeas o estadounidenses (51 contra 63 años), en parte debido a la estructura de la pirámide poblacional en la que predomina un mayor porcentaje de población joven.2
Oncogenómica
Una de las áreas de la biomedicina que más se han beneficiado de la caracterización del genoma ha sido la oncología, tanto para entender los mecanismos básicos de los procesos de transformación neoplásica, como para el desarrollo de nuevos servicios para un mejor pronóstico y evaluación del riesgo en pacientes oncológicos. Esto ha abierto una nueva área de investigación en oncología basada en la caracterización genómica de las neoplasias: la oncogenómica.3-7
El cáncer de mama constituye uno de los primeros ejemplos de la traducción de la investigación genómica a las aplicaciones clínicas. A partir de perfiles moleculares se ha conseguido una mejor clasificación de los tumores y el desarrollo de nuevos fármacos, así como nuevos productos que tienen ya una aplicación clínica e instrumentos de evaluación de riesgo y respuesta terapéutica.
Clasificación genómica del cáncer de mama
Los primeros trabajos que analizaron los cambios en los patrones de expresión génica en el tejido mamario se llevaron a cabo al comparar la expresión de 8 102 genes en 65 muestras quirúrgicas de cáncer de mama para obtener "firmas" o "fotografías" moleculares de cada tumor. Los resultados de dicho estudio evidenciaron la presencia de diversos fenotipos moleculares y ello sugirió la existencia de una gran diversidad biológica en los tumores mamarios:
1. El grupo ERBB2, que expresa altos niveles del gen ERBB2, así como de otros genes localizados en el amplicón ERBB2 (negativos al receptor de estrógenos). Estos tumores muestran una mejor respuesta a la quimioterapia y cerca de 50% responde al tratamiento con Trastuzumab.
2. El grupo "parecido al normal" se caracteriza por expresar un gran número de genes propios del epitelio mamario normal (negativos al receptor de estrógenos).
3. El grupo "basal" expresa genes característicos de las células basales de la mama, en particular queratinas 5 y 17, y son negativos al receptor de estrógenos y amplificación de ERBB2, es decir, son negativos triples. Este subtipo tiene el pronóstico más sombrío.
4. El grupo "luminar" se distingue por la expresión, relativamente alta, de muchos genes expresados en las células epiteliales de la luz de los conductos mamarios, incluido el receptor de estrógenos. Son tumores negativos a ERBB2 y suelen tener la mejor tasa de sobrevida.
Los análisis genómicos iniciales han demostrado que dentro de la denominación "tumores negativos a receptor de estrógenos" se encuentran al menos tres entidades biológicamente distintas (grupos 1 a 3), las cuales deberían tratarse como enfermedades diferentes.8,9 En un estudio posterior se analizaron 78 carcinomas mediante un microarreglo de 8 102 genes. Este trabajo permitió depurar la lista de genes capaces de diferenciar a los cuatro patrones previamente descritos y reducirla a 476 genes, algo que se conoce como perfil intrínseco de expresión. De esta forma, se han logrado relacionar perfiles de expresión genómica con la sobrevida general de las pacientes. En consecuencia, los subtipos ERBB2 y el basal parecen vincularse con una menor sobrevida. Asimismo, los tumores de los subgrupos luminar B y luminar C se identificaron como entidades clínicas diferentes con un curso más agresivo, en particular en relación con la reincidencia del tumor.10
De manera paralela, otro grupo de investigación analizó los patrones de expresión de unos 25 000 genes en 117 tumores de mama. Al aplicar una medida de clasificación supervisada, se identificó un patrón de 70 genes que resultó altamente predictivo para el desarrollo de metástasis distantes en un periodo de cinco años en pacientes sin evidencia de metástasis linfática regional. Este hallazgo indica que es posible llevar a cabo evaluaciones pronósticas del curso clínico en tumores de mama a partir del análisis de la lesión primaria. Este trabajo demuestra la utilidad de las firmas moleculares para detectar patrones de expresión que tienen un mayor valor predictivo que los parámetros clínicos tradicionales. Además, permite reconocer a los pacientes que se beneficiarán en mayor proporción del tratamiento adyuvante al analizar tumores primarios de mujeres jóvenes en estadios I y II (menores de 53 años).11,12
Además de la utilidad predictiva, la estadificación basada en patrones de expresión se correlaciona con la respuesta a la quimioterapia preoperatoria. En consecuencia, a través del análisis del tejido obtenido de biopsias por aspirado de aguja fina se determinó que los subtipos parecido al basal y ERBB2 son más sensibles al tratamiento preoperatorio con paclitaxel y doxorrubicina en comparación con los patrones parecido a normal y luminar.13
La utilidad clínica y la relevancia biológica de estos hallazgos también se demostraron al analizar los patrones de expresión de lesiones metastásicas, en las cuales se identificó un patrón de expresión muy similar al del tumor primario, aun si la metástasis distante se desarrollaba años después del diagnóstico y se encontraba en un microambiente distinto al del tumor primario.14,15
Diferentes perfiles de expresión y evaluación del riesgo
Para evaluar la correlación entre los resultados obtenidos con el perfil de genes intrínseco y la predicción del curso clínico con base en el patrón de expresión de 70 genes, se analizaron 115 tumores independientes, con agrupamiento jerárquico basado en los 534 genes que mejor diferenciaban a los subtipos luminar, basal, ERBB2 y parecido a normal. Este mismo análisis se aplicó a los datos, de los cuales se obtuvo el patrón predictivo de los 70 genes. Los resultados indicaron que, cualquiera que fuera la plataforma de microarreglos utilizada, se reproduce de forma consistente la identificación de los subgrupos de tumores en datos generados de forma independiente.
Además de corroborar la presencia de los subgrupos, este trabajo permitió correlacionar al subtipo de tumores con patrones de expresión basal y ERBB2 con el grupo de alto riesgo, definido por el patrón de 70 genes, mientras que los subtipos luminar A y luminar B mostraron una correlación con un buen pronóstico y un pronóstico intermedio, respectivamente.16
Dado que se han desarrollado diferentes tipos de patrones de expresión con capacidades predictivas en distintas cohortes de pacientes y cada grupo ha utilizado plataformas tecnológicas diferentes, la lista de genes específicos muestra poca sobreposición entre los distintos estudios. Esto puede impedir la aplicación clínica de tales hallazgos. El problema se analizó mediante la comparación de cinco patrones de expresión en una cohorte de 295 pacientes en relación con su capacidad de evaluar riesgo, al margen de los genes analizados en cada uno de lo perfiles. Se compararon cinco patrones: a) el modelo de progresión basado en 70 genes (MammaPrintTM);11,12 b) modelo de respuesta a heridas;17,18 c) puntaje de recurrencia (OncotypeDXTM);19,20 d) modelo de subtipos intrínsecos (luminar A, luminar B, ERBB, receptor de estrógenos negativo, basal y parecido a normal);8,16 y e) modelo de tasa de expresión de dos genes (relación de tasa de expresión entre HOXB13 e IL17BR).
Los resultados indican que la mayor parte de los modelos presenta niveles altos de concordancia en cuanto a su capacidad de predicción clínica en muestras individuales. Casi todos los tumores que se identificaron como basales, ERBB2-negativos a receptor de estrógenos o luminares B también se clasificaron en el grupo de mal pronóstico mediante los modelos de 70 genes, activación de respuesta a heridas y una elevada puntuación de recurrencia. El patrón de 70 genes y el modelo de puntuación de recurrencia concordaron en la clasificación de desenlace clínico en 77 a 81% de los casos, lo cual resulta relevante, dado que estas pruebas se aplican ya en el ámbito clínico. En el cuadro I se presentan algunas de las pruebas con base genómica para evaluar el riesgo de cáncer de mama.21
Correlación entre perfiles de expresión, clasificación molecular y patrones de alteración en el número de copias de DNA
El análisis de aberraciones en el número de copias de DNA en el cáncer de mama ha identificado regiones específicas de amplificaciones en 8q11, 1q21, 17q11, y 11q13, así como deleciones en segmentos que contienen genes supresores de tumor conocidos, como PTEN y CDKN2A. En términos comparativos, los tumores del subtipo basal presentan más alteraciones en el número de copias que los otros subtipos, lo que indica un mayor grado de inestabilidad cromosómica en este subtipo tumoral. Entre los sitios de interés de amplificación relacionados con peor pronóstico clínico se encuentran 8q33.3 (EDD1, WDSOF1), 8q24.11-13 (THRAP6, DCC1, SQLE, SPG8) y 11q14.1 (NDUFC2, ALG8, USP35). En realidad, la amplificación de cualquiera de estas regiones permite identificar a pacientes con una menor sobrevida y presentación de metástasis distantes al margen de otros parámetros clínicos.22 La figura 1 muestra un histograma de frecuencias con las regiones del genoma humano que sufren alteraciones en el número de copias en un mayor porcentaje de los tumores mamarios.
Los tumores "triple negativos"23 se caracterizan por la ausencia de expresión de receptores hormonales (estrógenos y progesterona), así como de HER2, y constituyen cerca de 15% del total de los tumores de mama. Este subtipo de neoplasias pertenece en su mayoría al subgrupo de expresión basal, el cual se distingue por un curso clínico muy agresivo.24 Se han identificado alteraciones específicas en el número de copias relacionadas con este tipo de tumor, como amplificaciones en 9p24-p21, 10p15-p13, 12p13, 13q31-q34, 18q12, 18q21-q23 y 21q22.
El uso de microarreglos de genotipificación de alta densidad ha identificado que los patrones de pérdida y ganancia de material genético son diferentes en subtipos tumorales clasificados mediante expresión. Estas diferencias incluyen la amplificación de 12q, para el tipo luminar B (gen RAB1B), la amplificación de 10p para el subtipo basal (gen KIAA1217) y la deleción recurrente de 16q en el subtipo luminar A. Esta deleción constituye un factor de buen pronóstico en todos los subgrupos tumorales.
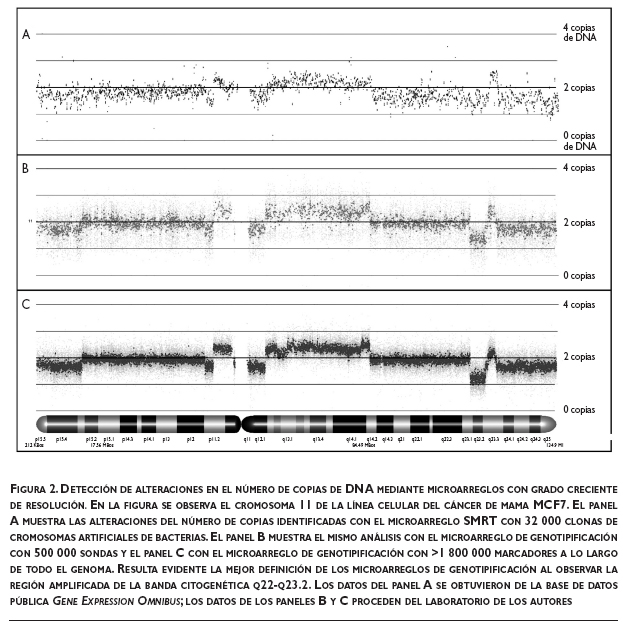
El uso del microarreglo de SNP capaz de identificar 500 000 variaciones genómicas, junto con el análisis de datos de expresión génica, permitieron determinar la importancia funcional de las alteraciones del número de copias de DNA. Estos análisis no sólo destacan la importancia de los genes identificados con anterioridad, como ERBB2, sino que además posibilitan una mejor definición de otras regiones importantes, como las amplificaciones en 8p12 y 11q13.5-q14.2. Esto resulta en una notoria reducción del número de genes blancos con posible relevancia biológica. Un ejemplo de esta utilidad es la reducción de la amplificación detectada con regularidad en 17q23.2 a una región de 249 kb que contiene al gen RPS6KB1 y al micro-RNA presumiblemente oncogénico mir-21.25 La figura 2 muestra la capacidad de detección de alteraciones en el número de copias de DNA de diferentes plataformas de microarreglos y ejemplifica la forma en que una mejor resolución hace posible identificar alteraciones genómicas más puntuales en el genoma de la célula neoplásica.
Micro-RNA y cáncer de mama
Estudios iniciales en cáncer de mama han demostrado que es posible diferenciar tejido normal de tumores mediante patrones de expresión de micro-RNA: los que presentan un mayor cambio son mir-125b, mir-145, mir-21 y mir-155.26
Asimismo, se ha demostrado que genes relevantes en la transformación neoplásica de la mama son susceptibles a la regulación por micro-RNA, como en el caso de ERBB2 y ERBB3,27 receptor de estrógenos,28 tropomiosina 129 y caderina E,30 entre otros. En fecha reciente se describió la función del micro-RNA-10b (miR-10b) en el inicio del proceso de invasión y metástasis a través de la inhibición de HOX-D10 y la sobreexpresión de RHOC.31 Estos datos sugieren que los micro-RNA, dada su capacidad de regular la función de una gran cantidad de blancos transcripcionales, podrían determinar los patrones de expresión a nivel de RNA mensajero en los diferentes subtipos de cáncer de mama. Esta hipótesis se confirma en trabajos recientes, que demuestran la presencia de micro-RNA específicos en diferentes subtipos tumorales, como el basal y el luminar.32 El papel de estas moléculas como posibles biomarcadores de riesgo se ha demostrado a través del análisis de miR-126 y miR-335. La expresión de estos micro-RNA se pierde en la mayoría de los tumores primarios de mama, sobre todo en los pacientes que presentan una recaída y la pérdida de la expresión se vincula con un menor tiempo de desarrollo de metástasis.33
Análisis de susceptibilidad genética y riesgo de cáncer de mama
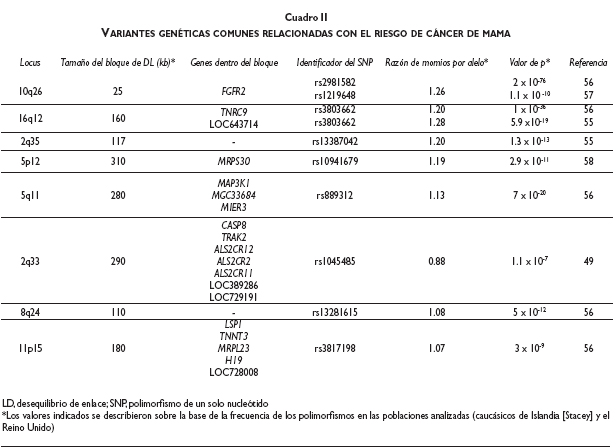
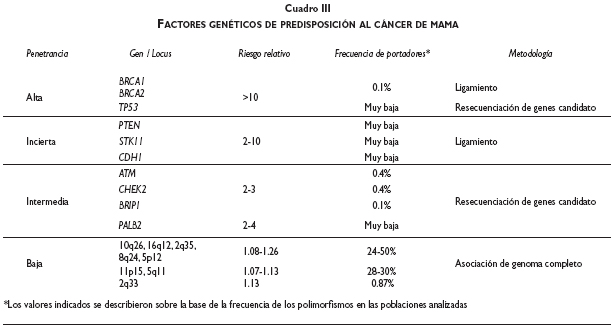
Los tres abordajes más importantes que se han aplicado para el descubrimiento de factores genéticos de predisposición al cáncer de mama son los estudios de ligamiento en el genoma completo, la búsqueda de mutaciones en genes candidato mediante secuenciación y, en fecha reciente, los estudios de asociación del genoma completo. Hoy día se conocen tres grupos de factores de predisposición genética al cáncer de mama, basados en el riesgo relacionado con cada grupo (cuadro II).
Estudios de ligamiento
El primer grupo de riesgo lo constituyen los genes de alta penetrancia relacionados con el cáncer de mama de tipo familiar; a través de estudios de ligamiento fue posible mapear los genes BRCA1 y BRCA2.34-37 Las mutaciones en BRCA1 y BRCA2 confieren alrededor de 15 a 20% del riesgo relativo familiar para cáncer de mama en la población caucásica.38
Las mutaciones en TP53, dentro del marco del síndrome de Li-Fraumeni, también se consideran como de alta penetrancia, ya que las pacientes con este síndrome presentan un riesgo significativamente más elevado a padecer cáncer de mama.39 También en los síndromes de Cowden (mutaciones en PTEN) y Peutz-Jeghers (STK11) y el síndrome de cáncer gástrico difuso hereditario (mutaciones en CDH1) se ha identificado una mayor susceptibilidad a desarrollar cáncer de mama. Sin embargo, las mutaciones en TP53, PTEN, STK11 y CDH1 son muy raras en los tumores de mama esporádicos, por lo que su contribución al riesgo relativo fuera del contexto de sus síndromes adjuntos es muy baja.40-44
Estudios de resecuenciación de genes candidato
Esta aproximación ha identificado mutaciones en genes como CHEK2, ATM, BRIP1 y PALB2, los cuales pueden elevar el riesgo relativo de padecer la enfermedad de dos a cuatro veces.45-48 Por otra parte, existe un polimorfismo codificante en el gen CASP8 que se vincula con una reducción moderada del riesgo de padecer cáncer de mama.49 Sin embargo, la frecuencia de estas variaciones en la población general es baja y, aun si se consideran todos los genes de susceptibilidad conocidos hasta la fecha, todavía queda una proporción de 70 a 75% de los casos de cáncer de mama que no presenta estas variantes genéticas.50
Estudios de asociación, genes candidato y genoma completo
Los estudios genómicos del cáncer de mama se llevaron a cabo de manera inicial a través de una estrategia de gen candidato.51 Desafortunadamente, debido a limitaciones en el número de muestras analizadas y su diseño, muchos de los resultados de estos estudios no fueron replicables.52 En fecha reciente ha sido posible el desarrollo de plataformas tecnológicas que permiten evaluar cientos de miles de polimorfismos de un solo nucleótido (SNPs) en grandes cantidades de muestras, lo cual abre la posibilidad de llevar a cabo estudios de asociación en todo el genoma y elimina el sesgo debido a preconcepciones acerca del papel de genes específicos en las enfermedades.53,54
En el caso del cáncer de mama se han efectuado cuatro estudios de asociación del genoma completo, que en conjunto han analizado cerca de 40 000 casos contra 79 000 controles (cuadro III).55-58 El diseño experimental de estos estudios se ha dividido en etapas, lo que hace más eficiente el análisis y reduce sus costos. Por ejemplo, el estudio de Easton analizó de manera inicial un grupo de 390 casos, ya sea con un historial de cáncer de mama hereditario o bien con tumores bilaterales, contra 364 controles mediante una plataforma de microarreglos de genotipificación para el análisis de 227 876 SNP. Para la segunda etapa se seleccionó 5% de los SNP más significativos (12 711), los cuales se genotipificaron en 3 990 casos y 3 916 controles independientes. En la tercera etapa se seleccionaron los 30 marcadores más significativos y se analizaron en 21 860 casos y 22 578 controles provenientes de 22 estudios internacionales.56 Las variantes genéticas reconocidas en estos estudios elevan el riesgo relativo de cáncer de mama en <1.5. Sin embargo, en virtud de los enormes números de casos y controles analizados, el diseño en etapas que permite la replicación en poblaciones independientes y la astringencia estadística de los análisis, el poder de estas asociaciones es muy elevado.
La señal de asociación más sólida detectada hasta ahora se localiza en la región 10q26 (cuadro III), en el intrón 2 del gen FGFR2. Este gen se ha encontrado mutado en diferentes tumores, si bien las bases moleculares acerca del modo en que esta variante incrementa el riesgo de padecer cáncer de mama se desconoce. La siguiente variante más relacionada se halla en la región 16q12, en un bloque de desequilibrio de ligamiento que contiene al gen TNRC9 y cuya expresión puede predecir la capacidad metastásica de los tumores primarios a hueso.59 La señal en 5p12 se encuentra en desequilibrio de ligamiento con el gen MRPS30, que codifica para una proteína de ribosomas mitocondriales que interviene en el proceso de apoptosis. Por otro lado, la variante situada en 5q11 se halla en el bloque de desequilibrio de ligamiento donde se localizan tres genes; el MAP3K1 es el candidato más interesante, ya que codifica a una cinasa activada por mitógenos que interviene en el control de señalización celular. En cuanto al locus 2q33, la región incluye el gen CASP8, participante en la apoptosis, aunque el bloque de desequilibrio de ligamiento también incluye a otros genes cuya función no se ha determinado en el cáncer.
La asociación con variantes en 8q24 se localiza en una región muy interesante, ya que diversos estudios la han encontrado vinculada con el cáncer de próstata60, colon61 y vejiga.62 A pesar de la cercanía con el oncogén C-MYC, las bases moleculares que explican cómo estas variaciones afectan la carcinogénesis todavía se desconocen. Por último, la región 11p15 situada dentro del intrón 10 del gen LSP1, que codifica a una proteína específica de linfocitos, también se ha relacionado con cáncer de mama.
Análisis recientes indican que algunos de los genes en los que se localizan dichos marcadores (TNRC9, FGFR2 y MAP3K1) presentan expresión diferencial en los distintos subtipos de tumores, definidos por perfiles de expresión.63 Asimismo, la presencia de algunas de estas variaciones se ha vinculado con positividad para receptores hormonales, bajo grado histológico, metástasis en ganglios y tiempo de sobrevida.64
Estudios de resecuenciación masiva en el cáncer de mama
En la actualidad, los métodos de secuenciación automatizada han dado un salto significativo, abatiendo los costos en grado notorio y, sobre todo, aumentado la capacidad de secuenciación en forma masiva. En el caso del cáncer de mama, las técnicas de secuenciación de nueva generación se han utilizado para identificar los sitios de unión del receptor de estrógenos alfa65 y aún no se ha notificado la secuencia completa del genoma de un tumor de mama. Sin embargo, mediante tecnología de secuenciación tipo Sanger, se analizó la secuencia de 20 857 transcritos provenientes de 18 191 genes en una muestra inicial de 11 tumores y dos tejidos normales del mismo paciente (grupo de descubrimiento).66 Los genes que se encontraron mutados en el grupo de descubrimiento se analizaron con posterioridad en un grupo independiente de 24 muestras (grupo de validación). Este análisis identificó un total de 506 mutaciones somáticas, de las cuales 79 se validaron en la muestra independiente y se distribuyeron en 62 genes. Estos genes codifican a proteínas que participan en 108 vías metabólicas, muchas de las cuales incluyen las vías de señalización de 3-cinasa de fosfatidilinositol (PI3K) y NF-κB.
Traducción de los hallazgos genómicos a la práctica clínica y salud pública
La traducción de resultados genómicos del cáncer de mama en productos clínicos se ha observado sobre todo en el campo de los perfiles de expresión de RNA. Tanto el patrón analizado por el modelo de 70 genes (MammaPrint) como la firma molecular de 21 genes (OncotypeDX) se utilizan en ensayos clínicos para determinar su utilidad y desempeño, en comparación con marcadores pronósticos tradicionales para determinar si se asigna quimioterapia adyuvante a pacientes de cáncer de mama. En el caso del MammaPrint, se efectúa el estudio MINDACT (Microarray In Node Negative Disease may Avoid Chemo Therapy) que compara el método tradicional para la evaluación de recurrencia del tumor con la firma molecular de 70 genes. Por otra parte, el ensayo clínico Trial Assigning IndividuaLized Options for Treatment (Rx), o TAILORx, analiza la utilidad de la firma basada en 21 genes. Las pacientes con diagnóstico reciente de cáncer de mama, con receptores de estrógeno o progesterona positivos y negativos para amplificación de Her2/neu, y cuyos tumores aún no metastatizan a los ganglios linfáticos, son elegibles para este estudio.
Los alelos de riesgo identificados mediante estudios de asociación de genoma completo continúan bajo evaluación. En fecha reciente se evaluó el perfil de riesgo generado con seis de estos alelos en cuanto a su capacidad de determinar los riesgos individual y poblacional en la población del Reino Unido. Los resultados indican que el perfil no es suficientemente poderoso para predecir el riesgo individualizado. Sin embargo, es capaz de estratificar a la población general con base en riesgos relativos de acuerdo con grupos de edad. De esta forma, una mujer con un perfil de riesgo genético determinado por los seis SNP podría ubicarse en un percentil de riesgo poblacional específico para calcular el riesgo relativo relacionado con su edad. Las mujeres de 50 años que estuvieran en el quinto percentil de la distribución de riesgo tendrían un riesgo a 10 años de 1.5%, mientras que las mujeres de 41 años, ubicadas en el percentil 95, tendrían un riesgo a 10 años de 2.3%.67
Otro aspecto relevante de la investigación en el cáncer de mama es la farmacogenómica. Los polimorfismos en genes como CYP2D6, CYP3A4, CYP3A5, CYP19A1 y SULT1A1 se han relacionado con la eficiencia clínica del tamoxifén y las aromatasas. Asimismo, las variantes de los genes CBR3, ABCB1 y genes de estrés oxidativo se vinculan con una respuesta a antraciclinas y una respuesta clínica general,68 lo cual los hace candidatos interesantes para su evaluación en la población mexicana.
Conclusiones y perspectivas
El avance de las tecnologías de análisis genómico ha permitido identificar firmas moleculares para una mejor predicción del riesgo y evaluar la respuesta al tratamiento en pacientes afectadas. Las pruebas que evalúan perfiles de expresión como herramientas de predicción de riesgo se utilizan ya en la práctica clínica con buenos resultados. Por otro lado, los estudios de asociación de genoma completo han identificado variantes genéticas comunes vinculadas con un incremento del riesgo relativo de padecer cáncer de mama, los cuales podrían utilizarse como herramientas de estratificación del riesgo poblacional en programas de salud pública. Sin embargo, la mayor parte de los factores genéticos que modifican el riesgo y la evolución del cáncer de mama aún está por descubrirse.
Además, casi todos los estudios en este campo se han llevado a cabo en poblaciones caucásicas que poseen una estructura poblacional distinta de la población mestiza mexicana y latinoamericana. Datos recientes indican que el grado de componente ancestral europeo en pacientes latinas se relaciona con un riesgo incrementado de cáncer de mama,69 lo que sugiere la presencia de diferencias genéticas posiblemente vinculadas con el desarrollo de esta enfermedad.
Esto hace evidente la necesidad de robustecer los programas de investigación científica multidisciplinarios tendientes a identificar la frecuencia de las alteraciones genómicas descritas en la población mexicana y latinoamericana, ya que en ellas se desconocen aspectos fundamentales, como la distribución de frecuencia de alelos de riesgo o la incidencia de los distintos perfiles de expresión génica en tumores de mama. Es necesario el desarrollo de programas de investigación en medicina genómica para ofrecer mejores oportunidades diagnósticas y terapéuticas, a través de la correlación de la información clínica y los hallazgos genómicos, lo cual conducirá a una práctica médica cada vez más personalizada que permita la determinación del riesgo a esta enfermedad, así como la predicción de la respuesta al tratamiento y su pronóstico. Su aplicación en los próximos años tendrá importantes repercusiones en la calidad de vida de las mujeres en riesgo y sus familias, al contribuir al desarrollo de programas de prevención más individualizados y, en su caso, con mayor eficacia terapéutica.
Referencias
1. Registro Histopatológico de Neoplasias Malignas [database on the Internet]. Secretaría de Salud. 2001 [cited 2007]. Disponible en: http://www.dgepi.salud.gob.mx/diveent/RHNM.htm. [ Links ]
2. Instituto Nacional de Estadística, Geografía e Informática. Estadísticas a propósito del día mundial contra el cáncer. México: INEGI, 2007. [ Links ]
3. Morozova O, Marra MA. From cytogenetics to next-generation sequencing technologies: advances in the detection of genome rearrangements in tumors. Biochem Cell Biol 2008; 86(2):81-91. [ Links ]
4. Gresham D, Dunham MJ, Botstein D. Comparing whole genomes using DNA microarrays. Nat Rev Genet 2008; 9(4):291-302. [ Links ]
5. Velculescu VE. Defining the blueprint of the cancer genome. Carcinogenesis 2008. [ Links ]
6. Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008; 452(7189):872-876. [ Links ]
7. Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet 2008; 24(3):133-141. [ Links ]
8. Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature 2000; 406(6797):747-752. [ Links ]
9. Lonning PE, Sorlie T, Perou CM, Brown PO, Botstein D, Borresen-Dale AL. Microarrays in primary breast cancer--lessons from chemotherapy studies. Endocr Relat Cancer 2001; 8(3):259-263. [ Links ]
10. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001; 98(19):10869-10874. [ Links ]
11. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415(6871):530-536.
12. van de Vijver MJ, He YD, Van't Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347(25):1999-2009.
13. Rouzier R, Perou CM, Symmans WF, Ibrahim N, Cristofanilli M, Anderson K, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res 2005; 11(16):5678-5685. [ Links ]
14. Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, Van't Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci USA 2003; 100(26):15901-15905. [ Links ]
15. Weigelt B, Hu Z, He X, Livasy C, Carey LA, Ewend MG, et al. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res 2005; 65(20):9155-9158. [ Links ]
16. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 2003; 100(14):8418-8423. [ Links ]
17. Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci U S A 2005; 102(10):3738-3743. [ Links ]
18. Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2004; 2(2):E7. [ Links ]
19. Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004; 351(27):2817-2826. [ Links ]
20. Paik S, Tang G, Shak S, Kim C, Baker J, Kim W, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol 2006; 24(23):3726-3734. [ Links ]
21. Ross JS, Hatzis C, Symmans WF, Pusztai L, Hortobagyi GN. Commercialized multigene predictors of clinical outcome for breast cancer. Oncologist 2008; 13(5):477-493. [ Links ]
22. Chin SF, Teschendorff AE, Marioni JC, Wang Y, Barbosa-Morais NL, Thorne NP, et al. High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol 2007; 8(10):R215. [ Links ]
23. Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol 2007; 8(3):235-244. [ Links ]
24. Abd El-Rehim DM, Ball G, Pinder SE, Rakha E, Paish C, Robertson JF, et al. High-throughput protein expression analysis using tissue microarray technology of a large well-characterised series identifies biologically distinct classes of breast cancer confirming recent cDNA expression analyses. Int J Cancer 2005; 116(3):340-350. [ Links ]
25. Haverty PM, Fridlyand J, Li L, Getz G, Beroukhim R, Lohr S, et al. High-resolution genomic and expression analyses of copy number alterations in breast tumors. Gen Chrom Can 2008; 47(6):530-542. [ Links ]
26. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 2005; 65(16):7065-7070. [ Links ]
27. Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem 2007; 282(2):1479-1486. [ Links ]
28. Adams BD, Furneaux H, White BA. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol 2007; 21(5):1132-1147. [ Links ]
29. Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 2007; 282(19):4328-4336. [ Links ]
30. Hurteau GJ, Carlson JA, Spivack SD, Brock GJ. Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 2007; 67(17):7972-7976. [ Links ]
31. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007; 449(7163):682-688. [ Links ]
32. Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin SF, Dunning MJ, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Bio 2007; 8(10):R214. [ Links ]
33. Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008; 451(7175):147-152. [ Links ]
34. Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990; 250(4988):1684-1689. [ Links ]
35. Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994; 265(5181):2088-2090. [ Links ]
36. Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378(6559):789-792. [ Links ]
37. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266(5182):66-71. [ Links ]
38. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian Breast Cancer Study Group. Br J Cancer 2000; 83(10):1301-1308. [ Links ]
39. Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene 2001; 20(34):4621-4628. [ Links ]
40. Lalloo F, Varley J, Moran A, Ellis D, O'Dair L, Pharoah P, et al. BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur J Cancer 2006; 42(8):1143-1150. [ Links ]
41. Bignell GR, Barfoot R, Seal S, Collins N, Warren W, Stratton MR. Low frequency of somatic mutations in the LKB1/Peutz-Jeghers syndrome gene in sporadic breast cancer. Cancer Res 1998; 58(7):1384-1386. [ Links ]
42. Nelen MR, Padberg GW, Peeters EA, Lin AY, van den Helm B, Frants RR, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet 1996; 13(1):114-116. [ Links ]
43. Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391(6663):184-187. [ Links ]
44. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature 1998; 392(6674):402-405. [ Links ]
45. Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002; 31(1):55-59. [ Links ]
46. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 2007; 39(2):165-167. [ Links ]
47. Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006; 38(8):873-875. [ Links ]
48. Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 2006; 38(11):1239-1241. [ Links ]
49. Cox A, Dunning AM, Garcia-Closas M, Balasubramanian S, Reed MW, Pooley KA, et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet 2007; 39(3):352-358. [ Links ]
50. Turnbull C, Rahman N. Genetic predisposition to breast cancer: past, present, and future. Annu Rev Genomics Hum Genet 2008; 9:321-345. [ Links ]
51. Daly AK. Candidate gene case-control studies. Pharmacogenomics 2003; 4(2):127-139. [ Links ]
52. Pharoah PD, Dunning AM, Ponder BA, Easton DF. Association studies for finding cancer-susceptibility genetic variants. Nat Rev Cancer 2004; 4(11):850-860. [ Links ]
53. Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science 1996; 273(5281):1516-1517. [ Links ]
54. Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest 2008; 118(5):1590-1605. [ Links ]
55. Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 2007; 39(7):865-869. [ Links ]
56. Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447(7148):1087-1093. [ Links ]
57. Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 2007; 39(7):870-874. [ Links ]
58. Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 2008; 40(6):703-706. [ Links ]
59. Smid M, Wang Y, Klijn JG, Sieuwerts AM, Zhang Y, Atkins D, et al. Genes associated with breast cancer metastatic to bone. J Clin Oncol 2006; 24(15):2261-2267. [ Links ]
60. Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 2007; 39(5):631-637. [ Links ]
61. Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 2007; 39(8):984-988. [ Links ]
62. Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet 2008; 40(11):1307-1312. [ Links ]
63. Nordgard SH, Johansen FE, Alnaes GI, Naume B, Borresen-Dale AL, Kristensen VN. Genes harbouring susceptibility SNPs are differentially expressed in the breast cancer subtypes. Breast Cancer Res 2007; 9(6):113. [ Links ]
64. Garcia-Closas M, Hall P, Nevanlinna H, Pooley K, Morrison J, Richesson DA, et al. Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. PLoS Genet 2008; 4(4):e1000054. [ Links ]
65. Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet 2007; 3(6):e87. [ Links ]
66. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318(5853):1108-1113. [ Links ]
67. Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med. 2008; 358(26):2796-2803. [ Links ]
68. Tan SH, Lee SC, Goh BC, Wong J. Pharmacogenetics in breast cancer therapy. Clin Cancer Res 2008; 14(24):8027-8041. [ Links ]
69. Fejerman L, John EM, Huntsman S, Beckman K, Choudhry S, Perez-Stable E, et al. Genetic ancestry and risk of breast cancer among U.S. Latinas. Cancer Res 2008; 68(23):9723-9728. [ Links ]
 Solicitud de sobretiros:
Solicitud de sobretiros:
Dr. Gerardo Jiménez-Sánchez
Instituto Nacional de Medicina Genómica.
Periférico Sur 4124,Torre Zafiro II, 6o
Piso, col. Jardines del Pedregal. 01900 México DF. México
Correo electrónico: gjimenez@inmegen.gob.mx
Fecha de recibido: 24 de noviembre de 2008
Fecha de aprobado: 8 de enero de 2009


{kind=link}
{kind=link}
{kind=link}