










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalud Pública de México
versión impresa ISSN 0036-3634
Salud pública Méx vol.44 no.3 Cuernavaca may./jun. 2002
Importancia de las prostaglandinas en la amibiasis hepática
Blanca Sánchez-Ramírez, Dra en C,(1) Patricia Talamás-Rohana, Dra en C.(2)
Sánchez-Ramírez B, Talamás-Rohana P.
Importancia de las prostaglandinas en la amibiasis hepática.
Salud Publica Mex 2002;44:247-257.
El texto completo en inglés de este artículo está disponible en: http://www.insp.mx/salud/index.html
Resumen
Las prostaglandinas son importantes mediadores inflamatorios, pero también desempeñan un papel importante como reguladoras de las funciones de los linfocitos y los macrófagos. La inoculación por vía intrahepática o intraportal de trofozoitos viables de Entamoeba histolytica en hámsteres se caracteriza por una rápida respuesta inflamatoria aguda, en la cual los trofozoitos amibianos se ven rodeados sucesivamente por leucocitos polimorfonucleares, linfocitos y macrófagos. La incapacidad de estas células para contrarrestar la invasión amibiana ha sido demostrada en varios estudios. La prostaglandina E2 (PGE2) tiene potentes efectos sobre las células de la respuesta inmune; su participación durante la formación del absceso hepático se reportó recientemente. En este artículo hacemos una revisión de los hallazgos de los últimos años en relación con el estudio de los mediadores bioquímicos de la inflamación durante la infección con E. histolytica, y su posible participación en el establecimiento de la respuesta
Palabras clave: Entamoeba histolytica; prostaglandinas; prostaglandina-endoperóxido sintasa; citocinas; inflamación
Sánchez-Ramírez B, Talamás-Rohana P.
The importance of prostaglandins in hepatic amebiasis.
Salud Publica Mex 2002;44:247-257.
The English version of this paper is available at: http://www.insp.mx/salud/index.html
Abstract
Prostaglandins are important mediators of inflammation; they also play a role in the regulation of both lymphocyte and macrophage functions. Hamster's liver lesions resulting from intraportal or intrahepatic inoculation of living Entamoeba histolytica trophozoites are characterized by an acute inflammatory response, where trophozoites are successively surrounded by polymorphonuclear leukocytes, lymphocytes, and macrophages. Incapability of these cells to counteract amebic invasion has been demonstrated in some studies. Prostaglandin E2 (PGE2) has potent effects on immune cells; its participation in amebic liver abscess has been reported recently. This paper presents a review of recent discoveries on biochemical mediators produced during inflammation due to Entamoeba histolytica infection, and their possible role in establishing the host's immune response. The English version of this paper is available at: http://www.insp.mx/salud/index.html
Key words: Entamoeba histolytica; prostaglandins; prostaglandin-endoperoxido synthase; cytokines; inflammation
La inflamación comprende una serie de eventos complejos que surgen como respuesta del tejido a una agresión local o sistémica (figura 1). La respuesta local resulta en el reclutamiento de células fagocíticas que se encargan de eliminar el material endógeno o exógeno causante del daño. La respuesta sistémica puede alterar el micromedio ambiente para facilitar que este proceso se lleve a cabo. Debido a la cantidad de mediadores bioquímicos que participan en este proceso y la cantidad de células sobre las cuales influyen, se requiere de una estricta regulación para su control efectivo. Los cambios en el flujo sanguíneo, debidos a la vasodilatación del músculo liso, y las alteraciones en la permeabilidad vascular, promovidas por la contracción del citoesqueleto de las células endoteliales, favorecen la migración de leucocitos fagocíticos al sitio de la lesión (quimiotaxis). A su vez, la fagocitosis promueve la eliminación de microrganismos y la digestión de detritus celulares causantes de la inflamación, lo que permite la proliferación de las células del tejido conectivo y la reparación de la matriz extracelular. La inflamación puede iniciarse a partir de muy diversas vías: traumas en el tejido, la presencia de complejos inmunes, productos derivados de bacterias, virus o parásitos e, incluso, por la actividad de células tumorales o fagocíticas. La progresión de la inflamación de su fase aguda a crónica es importante, ya que ésta puede dañar el tejido del huésped a través de la producción, por los fagocitos,1 de proteasas y radicales del oxígeno.

En el caso de la infección hepática por Entamoeba histolytica, agente causal de la amibiasis, los hallazgos en necropsias señalaron la ausencia relativa de inflamación,2 motivo por el cual se asoció el daño encontrado en el tejido hepático con la actividad citolítica del parásito. No obstante, las evidencias obtenidas de estudios realizados en animales experimentales susceptibles a la infección hepática, ya sea por inoculación intraportal o intrahepática, han demostrado que el contacto de los trofozoitos amibianos con el tejido hepático promueve una fuerte reacción inflamatoria.3,4 Asimismo, estudios de ultraestructura de los primeros eventos de invasión de las amibas a la mucosa intestinal, revelaron la presencia de infiltración inflamatoria con lisis de leucocitos polimorfonucleares (PMNs), la cual fue atribuible tanto a las bacterias existentes en el lumen intestinal como a las amibas.5 Finalmente, el uso del modelo de asa cecal lavada y cerrada, permitió observar que el reclutamiento y la lisis de PMNs, así como el daño a la pared intestinal, pueden ocurrir en ausencia de la flora bacteriana y que el principal elemento citolítico en este estudio fueron los trofozoitos amibianos.6 Adicionalmente, la capacidad de los trofozoitos de E. histolytica para inmovilizar y destruir células, sobre todo PMNs, linfocitos y macrófagos, ha sido ampliamente demostrada in vitro.7,8
Sin embargo, aunque varios estudios fueron concluyentes en cuanto a la incompetencia de los PMNs para eliminar los trofozoitos amibianos, el papel de los mediadores inflamatorios liberados por estas células y otras, presentes en el tejido hepático, así como su efecto en el establecimiento de la respuesta inmune del huésped, no se contempló hasta hace unos años.
Panorámica del metabolismo del ácido araquidónico
La síntesis de los mediadores inflamatorios se puede resumir en tres fases (figura 2): 1) movilización del ácido araquidónico (AA) desde los fosfolípidos de la membrana celular, mediada por la actividad de la enzima fosfolipasa A2 (PLA2); 2) la conversión a endoperóxidos de prostaglandina PGG2 y PGH2, mediante la acción de una enzima ciclooxigenasa (COX, también denominada prostaglandin endoperóxido sintasa); y 3) la isomerización o reducción del endoperóxido PGH2 hacia una serie de derivados biológicamente importantes, como son las prostaglandinas de la serie E (PGE2), D (PGD2), F (PGF2a), prostaciclina (PGI2) y tromboxanos (TXA2), lo cual requiere la acción de enzimas sintasas específicas. El AA libre también puede ser movilizado hacia la producción de leucotrienos (LTs) y otros ácidos hidroxieicosatetranoicos (HETEs), a través de la acción de una enzima lipooxigenasa (LO).9 Todos estos compuestos contribuyen a la formación del exudado vascular debido al efecto vasodilatador de las PGs y al aumento del infiltrado celular mediado por la actividad quimiotáctica de los LTs del tipo B4 (LTB4).1
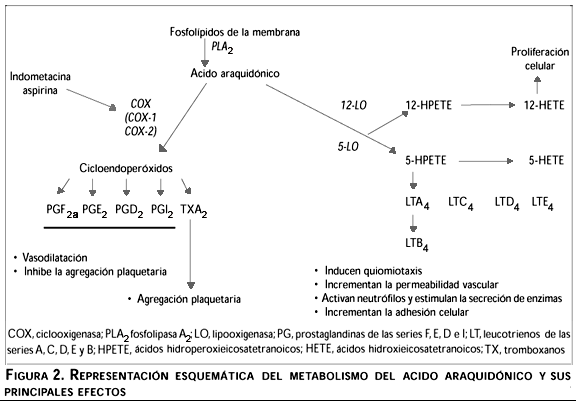
Se ha demostrado la existencia de al menos dos isoformas de la enzima COX: una forma constitutiva denominada COX-1, la cual participa en el mantenimiento de las condiciones fisiológicas homeostáticas; y una forma inducible denominada COX-2.10 COX-1 es una enzima glicoproteica con peso molecular de 70 kDa; se expresa en forma diferencial en las diversas células y tejidos de los mamíferos; su regulación obedece principalmente a la actividad hormonal y se le localiza en el nivel del retículo endoplásmico.11,12 En contraste, la expresión de la enzima COX-2 parece estar limitada a unos cuantos tipos celulares como macrófagos,13 fibroblastos,14 células endoteliales,11 células de Kupffer15 y, bajo ciertos estímulos, PMNs16 y linfocitos T.17 Niveles relativamente bajos del mRNA para COX-1 y COX-2 se han demostrado en varios tejidos mientras que, bajo la acción de agonistas específicos, los niveles de expresión del mRNA de COX-2 se incrementan en forma dramática y selectiva.11 Estas enzimas presentan una homología aproximada de 60% (considerando las variaciones entre especie) de acuerdo con las secuencias de aminoácidos. Las principales diferencias se localizan en los extremos amino y carboxilo terminal, por lo cual los sitios de acetilación para la hCOX-1 (Ser 529) y hCOX-2 (Ser 516), ambos cercanos al extremo carboxilo terminal, son sitios altamente conservados, de lo cual se desprende que ambas enzimas sean susceptibles a la inhibición mediante el uso de agentes antinflamatorios no esteroideos (NSAIDs) como la aspirina, la indometacina (Indo) y fluribuprofeno. A la fecha se han reportado nuevos compuestos con actividad antinflamatoria específica para cada una de las isoenzimas.9 Entre los agonistas encontrados para COX-2 se pueden mencionar citocinas (IL-1), mitógenos y factores de crecimiento (factor de crecimiento derivado de plaquetas, PDGF), lipopolisacáridos (LPS), ésteres de forbol (PMA, TPA) e inclusive la propia PGE2, por lo cual esta isoforma parece estar más involucrada con la generación y el mantenimiento de la respuesta inflamatoria.11,13-17
Hallazgos recientes reportan la existencia de diferentes enzimas sintasas de PGE2 acopladas a cada isoenzima COX.18 La enzima prostaglandina E sintasa citosólica (cPGES), también denominada p23, es expresada en forma constitutiva y ubicua en el citosol de casi todas las células y tejidos, y está asociada predominantemente con la actividad de COX-1. En tanto que la enzima prostaglandina E sintasa microsomal (mPGES o PGES-2), es una enzima perinuclear fuertemente asociada con células relacionadas con la respuesta inflamatoria, y en cuya funcionalidad se observó una marcada preferencia por COX-2.18
Efectos inmunomoduladores de la PGE2
Los derivados del AA actúan sobre muchos tipos de células y sus funciones son, por lo tanto, demasiado amplias para abarcarlas en su totalidad. Los efectos de la PGE2 son de particular importancia, debido a su acción sobre las células de la respuesta inmune, en particular sobre linfocitos y macrófagos. En el cuadro I se resumen algunos de los principales efectos sobre estos tipos celulares.

Adicionalmente, existe una estrecha relación entre las actividades de la PGE2 y algunos otros factores de la respuesta inmune, como son el óxido nítrico (NO) y el factor de necrosis tumoral (TNF-a). Se ha reportado que la PGE2 tiene la capacidad de regular la expresión de la enzima óxido nítrico sintasa (inducible) en forma dosis dependiente, ya que en líneas celulares de macrófagos J774 los LPS, en bajas dosis, inducen concentraciones insignificantes de PGE2 endógena, con secreción de NO; en tanto que, a dosis altas, los LPS generan niveles elevados de PGE2 que inhiben la liberación de NO.29 Asimismo, altos niveles de PGE2 bloquean la producción de TNF-a y generan la acumulación del mRNA del TNF-a, lo cual a su vez podría ejercer efectos negativos sobre la síntesis de NO.30
Por otra parte, la expresión de las moléculas del complejo mayor de histocompatibilidad clase II (MHC-II) en la superficie de los macrófagos de roedor, es un proceso altamente regulado dada su relevancia en la respuesta inmune y en la inflamación.31 La expresión de esta molécula es inducida por factores como el IFN-g, mientras que algunos agentes como el IFN-ab, los LPS y las PGE2 tienen la capacidad de inhibir el efecto inductor del IFN-g; en el caso particular de la PGE2 se inhibe tanto la expresión de las moléculas MHC-II, como la transcripción del gen,32 lo cual limita la capacidad cooperadora del macrófago.
Los macrófagos son la principal fuente de prostaglandinas en el sitio de la inflamación. Aunque éstas tienen una vida media muy corta, sus efectos in vivo se manifiestan principalmente en forma autocrina y paracrina sobre las células presentes en la lesión. Al ser la enzima COX-2 inducible, puede ser regulada por varios factores y varios tipos celulares presentes durante la inflamación, tal es el caso de la IL-4, la IL-10 y la IL-17. Aunque las primeras se consideran citocinas antinflamatorias, recientemente se descubrió que parte de estos efectos se deben a que ambas inhiben la producción de citocinas proinflamatorias como la IL-1 (a y b), el TNF-a, la IL-6 y la IL-8.16 Adicionalmente, incrementan la producción del receptor antagonista de la IL-1 (IL-1ra), el cual posee per se un efecto antinflamatorio.33 Asimismo, la IL-4 y la IL-10 inhiben la producción de PGE2 a nivel de la expresión del mRNA de COX-2 en neutrófilos,16 y en macrófagos humanos,33 en los cuales también se detectó un efecto similar por acción de la IL-13.34 Por su parte, la IL-17, una citocina descrita recientemente35 y la cual es producida únicamente por células T de memoria, muestra una actividad estimuladora sobre diversos tipos celulares como los macrófagos,36 fibroblastos37,38 y sinoviocitos.38 Al respecto se ha demostrado que la IL-17 estimula la producción de TNF-a, IL-1b, IL-6, IL-10, IL-12, PGE2 e IL-1ra en macrófagos,36 asimismo, induce la producción de IL-6, IL-8, factor estimulador de colonias de granulocitos (G-CSF), incrementa la expresión de la molécula de adhesión intracelular 1 (ICAM-1) y la producción de PGE2 en sinoviocitos y fibroblastos humanos.35,38 La desactivación de ese proceso estimulador mediado por IL-17, se lleva a cabo muy probablemente gracias a los efectos de la IL-4 y la IL-10, ya que estas citocinas revierten la acción estimuladora de la IL-17 en cuanto a la producción de IL-1b y de TNF-a.36,38 Adicionalmente, el TNF-a incrementa la capacidad de producción de PGE2 a través de la expresión del gen y de la proteína de COX-2.39 Por el contrario, el IFN-g, una citocina del subgrupo cooperador Th1, carece de efecto sobre la expresión de COX-2.34
Evidencias sobre los efectos inmunomoduladores de la PGE2 en modelos in vivo han sido encontradas en diversos tipos de infecciones, tanto por bacterias como por parásitos. Estos estudios han demostrado que los macrófagos de ratones infectados con Mycobacterium intracellulare producen altos niveles de PGE2, lo cual suprime su actividad, inhibiendo la liberación de citocinas específicas.40 De igual manera, se ha demostrado que el tratamiento con Indo, IFN-g, o una combinación de ambos, contribuye a la resolución de la infección tanto por Mycobacterium intracellulare40 como por Brucella abortus,41 y que en todos los casos se observa el restablecimiento de la actividad de los macrófagos. En el caso de infecciones por levaduras se ha descrito que Candida albicans tiene la capacidad de estimular la producción de eicosanoides (principalmente PGI2) en células endoteliales,42 la cual participa en la regulación de la respuesta inmune contra este patógeno. Además, recientemente se demostró la capacidad del Cryptococcus neoformans y de la Candida albicans para producir PGE2.43 En el caso de los parásitos, diversos estudios han revelado que una gran cantidad de ellos tienen la capacidad de producir PGE2,44 la cual es utilizada para regular la respuesta inmune del huésped y favorecer el proceso de invasión por el parásito.45-47
Las prostaglandinas y la respuesta inmune en la amibiasis
En el curso de la infección amibiana existe un estado de inmunosupresión que se caracteriza por la incapacidad del macrófago para inhabilitar y destruir al parásito. Este estado de anergia puede ser regulado por el parásito o bien, ser el resultado de los propios mecanismos de defensa del huésped. Debido a la invasión del tejido por el parásito son liberados al medio ambiente local una variedad de mediadores locales del tipo del TNF, la IL-1, probablemente la IL-6 y el factor activador de plaquetas (PAF). La acción combinada de estos factores causa fiebre y leucocitosis, por lo cual se ven involucrados en la generación de la sintomatología de la infección.48 Algunos de estos compuestos son potentes activadores de la enzima PLA2 y generan mediadores, como el mismo PAF, PGs, LTs, TXs, y metabolitos reactivos del oxígeno, además de moléculas con actividad bactericida o enzimas lisosomales, influyendo en forma importante sobre el sistema inmune.49 Aunque la liberación de estos agentes juega un papel básicamente protector para la homeostasis del tejido,50 puede contribuir al establecimiento de ciertos agentes infecciosos.
En la infección hepática con E. histolytica la liberación de estos factores probablemente sea la causa de la enorme concentración de células inflamatorias, sobre todo PMNs, que rodean al parásito desde los primeros minutos de su instalación en el tejido hepático. Algo similar ocurre durante la infección intestinal por este parásito, en la cual la mucosa intestinal presenta inflamación y acumulación de PMNs en la lámina propia. Posterior a la depleción de la capa de moco los trofozoitos se adhieren a las células epiteliales y, previa lisis de las mismas, proceden a invadir el tejido.50
La producción de citocinas proinflamatorias del tipo de la IL-8 ha sido estudiada in vitro, utilizando la línea celular T84 de adenocarcinoma de colon. En este modelo se demostró que la interacción del parásito con componentes de secreción, proteínas solubles y el contacto célula–célula, induce la producción de altos niveles del mRNA de la IL-8.51 Adicionalmente, la producción de IL-1 e IL-8 por células del epitelio intestinal, en respuesta a la interacción con trofozoitos y proteínas del parásito, ha sido confirmada in vivo e in vitro.52,53 La naturaleza de los factores amibianos que inducen la producción de estas citocinas no ha sido aclarada en su totalidad. Sin embargo, McGowan y colaboradores54 comprobaron que extractos proteicos de trofozoitos amibianos inducen cambios en el transporte de electrolitos en la mucosa intestinal, y que éstos están relacionados con la producción de PGE2, ya que el uso de inhibidores de la enzima COX (Indo y piroxicam) anula parcialmente el efecto del extracto amibiano.54 Esto sugiere que la inducción de la actividad de la enzima COX (cualquiera de las dos isoenzimas) y la consecuente producción de PGE2, podrían ser en parte inducidas por la IL-1, la IL-8, o bien por proteínas del parásito. Sin embargo, trabajos concluyentes a este respecto no se conocen.
Por otro lado, la infección con E. histolytica induce la producción de anticuerpos del tipo IgG, IgA e IgM contra un repertorio variable de antígenos. El papel de estos anticuerpos in vivo como parte de los mecanismos de defensa no es claro e, incluso, se sugiere que éstos podrían contribuir a la presentación de fenómenos de inmunosupresión,55 hipersensibilidad y daño por depósito de complejos inmunes.56,57 Adicionalmente, estudios de inmunización realizados con la subunidad de 170 kDa de la lectina de adherencia inhibible por galactosa mostraron una eficiencia de 67% en la prevención de la formación de absceso hepático amibiano en modelos experimentales, sin encontrarse correlación entre los títulos de anticuerpos desarrollados contra la lectina y la inmunidad protectora.58 Como criterio diagnóstico, la respuesta humoral tampoco es muy relevante, ya que ni la positividad ni el título de anticuerpos muestran correlación con la severidad de la infección, y no es posible discriminar entre un contacto reciente o una respuesta de memoria.59 La relación entre los niveles e isotipos de anticuerpos que se producen en el huésped durante la infección con E. histolytica y los niveles de PGE2, no ha sido descrita hasta ahora.
En cuanto a la respuesta inmune celular, los primeros estudios sobre la actividad de los macrófagos contra antígenos de E. histolytica se hicieron en leucocitos de pacientes con absceso hepático amibiano; los resultados mostraron una disminución en la producción de factor inhibidor de la migración (MIF), mismo que se restableció después del tratamiento y recuperación del paciente.60 Estudios sobre amibiasis experimental, efectuados en modelos animales, mostraron una baja respuesta en pruebas de hipersensibilidad tardía, aun en aquellos que presentaron altos títulos de anticuerpos.61 Adicionalmente, al estimular la producción de MIF en los macrófagos se aborta el estado de inmunosupresión, lo cual sugiere que la respuesta podría ser modulada por el parásito o por mecanismos intrínsecos al huésped; además, linfocitos de sangre periférica, células de bazo y macrófagos peritoneales de hámsteres inmunizados, poseen actividad amebicida in vitro,60,62-64 mientras que in vivo son incapaces de eliminar al trofozoito. La presencia de ciertos factores en el suero de pacientes con amibiasis hepática puede suprimir la actividad de macrófagos de individuos normales contra el parásito in vitro,65 la naturaleza de estos "factores" no ha sido descrita a la fecha. Por otra parte, la relación entre la anergia de los macrófagos y el incremento de la susceptibilidad ha sido demostrada en modelos de amibiasis hepática en ratones atímicos,66 depletados de macrófagos con antisueros específicos, o mediante el uso de silica.67 Finalmente, estudios hechos en jerbos mostraron que los macrófagos presentes en el absceso hepático amibiano se encuentran fuertemente inhibidos en su potencial cooperador y en sus funciones efectoras, caracterizadas por deficiencias en la generación del estallido respiratorio, una baja producción de reactivos intermediarios del oxígeno y una baja respuesta a citocinas inflamatorias, como la IL-1.68
La participación de los metabolitos del AA en los mecanismos que bloquean la activación de los macrófagos fue posteriormente demostrada por varios estudios en jerbos. Se observó que los macrófagos aislados del absceso hepático (AMos), y macrófagos peritoneales estimulados con extractos proteicos de E. histolytica, o interaccionados con el parásito, producen concentraciones elevadas de PGE2 y LTC4.69 Adicionalmente, se ha demostrado que el decremento en la producción de TNF en AMos involucra un mecanismo mediado por PGE2, ya que el pretratamiento de las células con Indo o con IFN-g restablece la producción de TNF.70 En forma similar, se ha presentado evidencia de que la PGE2 está involucrada en una disminución de la expresión del mRNA y de la síntesis de moléculas del MHC-II en el ratón.71 Estudios específicos sobre la actividad de E. histolytica y sobre la expresión de algunos genes, mostraron un incremento en la expresión del TNF-a, IL-1a/b y c-fos en macrófagos de médula ósea por exposición de éstos a extractos crudos de trofozoitos amibianos.70,72
Estudios que utilizaron como modelo el hámster, mostraron que el proceso de formación del absceso hepático amibiano cursa con una elevación significativa de los niveles de PGE2 plasmática.73 La relación entre el incremento de PGE2 plasmática y el patrón Th1 o Th2 en este modelo no se ha establecido a la fecha. Sin embargo, la inhibición de la síntesis de PGs mediante el tratamiento de los hámsteres con Indo redujo en 30 y 18% el peso del absceso y del hígado, respectivamente. La actividad de la enzima COX en fracción microsomal, obtenida de porciones lejanas (aparentemente sanas) y cercanas al absceso de animales infectados tratados o no con Indo, mostró una actividad menor en las zonas cercanas y un incremento de 150% en las zonas alejadas, con respecto al tejido hepático normal sin infectar.73 Estos resultados permiten sugerir que un incremento en la actividad de COX-2, inducida por el parásito durante el proceso de invasión, podría favorecer el proceso inflamatorio; interesantemente, reportamos recientemente que AMos expresan la enzima COX-2.74 Estudios, mediante inmunohistoquímica, en cortes de hígado de hámster infectado, revelaron que, tanto los macrófagos adyacentes a los trofozoitos amibianos, como aquellos macrófagos presentes en los espacios portales de zonas lejanas al absceso hepático, y en ausencia de trofozoitos, expresan la enzima COX-2.74 Los trofozoitos amibianos también expresaron en forma constante la presencia de COX-2, lo cual coincide con informes anteriores sobre la presencia de una enzima ciclooxigenasa y la producción de PGE2 por E. histolytica.44,75 Estos resultados apoyan la posibilidad de que las PGs estén involucradas en el mantenimiento del proceso inflamatorio y que participen en la inmunosupresión de la actividad del macrófago, así como en la evasión de la respuesta inmune. Sin embargo, se desconoce si la expresión de la enzima es dependiente del contacto macrófagos-trofozoitos, o si es inducida por los productos de secreción del parásito, ya sean Ags específicos, o producto de la actividad de la PLA2.
Por otra parte, diversas vías metabólicas llevan a la liberación de AA, entre otras, la actividad de la PLA2, considerándose esta última como la de mayor importancia en la regulación de la biosíntesis de PGs en el macrófago.76 La síntesis de novo de la COX-2 en diferentes tipos celulares tiene correlación con el incremento en la producción de PGE2, mientras que en otros tipos celulares este incremento está asociado con un aumento en la actividad de la PLA2. En E. histolytica se han reconocido dos enzimas fosfolipasas de tipo A, las cuales están involucradas en los mecanismos de citólisis de las células blanco por el parásito.77,78 La posible participación de las enzimas PLA2 de E. histolytica en los mecanismos de inducción de COX-2, para incrementar la biosíntesis de PGE2, aún no ha sido estudiada.
Adicionalmente, un gran número de estudios han demostrado que los metabolitos derivados del oxígeno (O2- y H2O2) y moléculas efectoras no oxidativas del tipo del TNF-a y NO se encuentran regulados durante la infección con este parásito. Prueba de ello es que in vitro la activación de macrófagos, mediada por citocinas, puede culminar en la muerte del parásito a través de mecanismos dependientes de H2O2 y proteasas. Sin embargo, macrófagos tratados con extractos de proteínas amibianas y posteriormente activados con IFN-g o LPS, presentaron un decremento significativo en su citotoxicidad, acompañado de una disminución en la producción de TNF-a y de los niveles de expresión del mRNA para la óxido nítrico sintasa (iNOS), con respecto a aquellos que no tuvieron contacto con las proteínas amibianas. La inhibición de la producción de PGE2 aumentó la producción de TNF-a y la capacidad citolítica de los macrófagos, sin embargo, no tuvo efecto sobre los niveles de expresión de iNOS.79 Estudios posteriores revelaron que la producción del NO, y la expresión de iNOS en los macrófagos interaccionados con proteínas de E. histolytica, es regulada por un mecanismo mediado por el factor transformante de crecimiento b1 (TGF-b1).80 El TGF-b1 se ha asociado con una regulación negativa de las funciones del macrófago, tanto in vivo como in vitro. Esta citocina disminuye la producción de otras monocinas como IL-1 y la IL-6, reduce la producción de H2O2 y de NO en macrófagos estimulados con LPS y ésteres de forbol, y menoscaba la actividad citolítica de los macrófagos sobre amastigostes de Leishmania major y Trypanosoma cruzi, y esquistosomulas de Schistosoma mansoni.81 Hasta hoy no se ha demostrado que la PGE2 o algún otro derivado del AA, esté involucrado en la regulación del TGF-b1.
Por otra parte, los antígenos de E. histolytica que inducen la producción de eicosanoides no han sido identificados o purificados. En resumen, podemos concluir que los niveles de PGs, producidos por las células inflamatorias del huésped (principalmente por la estimulación de COX-2) estimuladas por los antígenos del parásito (ya sean PLAs o las mismas PGs), pueden regular en forma negativa la actividad de los macrófagos locales y mermar su capacidad citolítica sobre el parásito. En la figura 3 se representan algunos de los principales elementos involucrados en la producción de PGE2 durante la infección hepática con E. histolytica. Los LTs, que son considerados como mediadores proinflamatorios, pueden a través de su actividad quimiotáctica, participar en el reclutamiento de un enorme número de leucocitos PMNs. En consecuencia, los PMNs pueden atraer a un mayor número de macrófagos hacia los sitios de invasión, en los cuales existe un medio ambiente supresor dada la alta concentración de PGs (ya sean derivadas del huésped o del parásito). La continuación de este proceso y otros metabolitos del AA en el reclutamiento y quimiotaxis de PMNs hacia el sitio de la lesión, la participación de estos elementos en el establecimiento de la respuesta inmune celular y, en particular, en el establecimiento de una respuesta de tipo Th2, son entre otras, preguntas que están por aclarar.

La caracterización de las COXs y de las moléculas que intervienen en su activación, así como la función que estas enzimas y los eicosanoides derivados de ellas tienen en la fisiología del parásito, pueden ser la clave para el desarrollo de inhibidores específicos que contribuyan al desarrollo de una nueva terapéutica de la amibiasis hepática.
Referencias
1. Evans SW, Whicher JT. An overview of the inflammatory process. En: Whicher JT, Evans SW, ed. Biochemistry of inflammation. Londres: Kluwer Academic Publishers, 1992:1-17. [ Links ]
2. Councilman WT, Lafleur HA. Amoebic dysentery. Johns Hopkins Hosp Rep 1891;2:395-548. [ Links ]
3. Tsutsumi V, Mena-López R, Anaya-Velázquez F, Martínez-Palomo A. Cellular bases of experimental amebic liver abscess formation. Am J Pathol 1984; 117:81-91. [ Links ]
4. Chadee K, Meerovitch E. The pathogenesis of experimentally induced amebic liver abscess in gerbil (Meriones unguiculatus). Am J Pathol 1984; 117:71-80. [ Links ]
5. Griffin JL. Human amebic dysentery. Electron microscopy of Entamoeba histolytica contacting, ingesting, and digesting inflammatory cells. Am J Trop Med Hyg 1972;21:895-906. [ Links ]
6. Martínez-Palomo A, Tsutsumi V, Anaya-Velázquez F, González-Robles A. Ultraestructure of experimental intestinal invasive amebiasis. Am J Trop Med Hyg 1989;41(3):273-279. [ Links ]
7. Guerrant RL, Brush J, Ravdin JI, Sullivan JA, Mandell GL. Interaction between Entamoeba histolytica and human polymorphonuclear neutrophils. J Infect Dis 1981;143:83-93. [ Links ]
8. Salata RA, Ravdin JI. Review of the human immune mechanisms directed against Entamoeba histolytica. Rev Infect Dis 1986;8(2):269-273. [ Links ]
9. Vane JR, Botting RM. A better understanding of anti-inflammatory drugs based on isoforms of cyclooxygenase (COX-1 and COX-2). Adv Prostaglandin Thromb Leuk Res 1995;23:41-48. [ Links ]
10. Holtzman MJ, Turk J, Shornick LP. Identification of a pharmacologically distinct prostaglandin H synthase in cultured epithelial cells. J Biological Chem 1992;267:21438-21445. [ Links ]
11. O'Neill GP, Ford-Hutchinson AW. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett 1993;330:156-160. [ Links ]
12. Spencer AG, Woods JW, Arakawa T, Singer II, Smith WL. Subcellular localization of prostaglandin endoperoxide H synthases 1 and 2 by immunoelectron microscopy. J Biol Chem 1998;273:9886-9893. [ Links ]
13. O'Sullivan MG, Huggins Jr EM, Meade EA, DeWitt DL, McCall CE. Lipopolysaccharide induces prostaglandin H synthase-2 in alveolar macrophages. Biochem Biophys Res Commun 1992;187:1123-1127. [ Links ]
14. Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 1991;266:12866-12872. [ Links ]
15. Zhang F, Warskulat U, Wettstein M, Schreiber R, Henninger HP, Decker K et al. Hyperosmolarity stimulates prostaglandin synthesis and cyclooxygenase-2 expression in activated rat liver macrophages. Biochem J 1995;312:135-143. [ Links ]
16. Niiro H, Otsuka T, Izuhara K, Yamaoka K, Ohshima K, Tanabe T et al. Regulation by interleukin-10 and Interleukin-4 of cyclooxygenase-2 expression in human neutrophils. Blood 1997;89:1621-1628. [ Links ]
17. Iñiguez MA, Punzón C, Fresno M. Induction of cyclooxygenase-2 on activated T lymphocytes: Regulation of T Cell activation by cyclooxygenase-2 inhibitors. J Immunol 1999;163:111-119. [ Links ]
18. Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem 2000;275(42):32783-32792. [ Links ]
19. Schad V, Phipps RP. Prostaglandin E2-dependent induction of B cells unresponsiveness. J Immunol 1989;143:2127-2132. [ Links ]
20. Roper RL, Brown DM, Phipps RP. Prostaglandin E2 promotes B lymphocyte Ig isotype switching to IgE. J Immunol 1995;154:162-170. [ Links ]
21. Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol 1991;146:108-113. [ Links ]
22. Hilkens CM, Vermeulen H, Joost van Neerven RJ, Snijdewint FGM, Wierenga EA, Kapsenberg ML. Differential modulation of T helper type 1 (Th1) and T helper type 2 (Th2) cytokine secretion by prostaglandin E2 critically depends on interleukin-2. Eur J Immunol 1995;25:59-63. [ Links ]
23. Anastassiou ED, Paliogianni F, Balow JP, Yamada H, Boumpas DT. Prostaglandin E2 and other cyclic AMP-elevating agents modulate IL-2 and IL2Ra gene expression at multiple levels J Immunol 1992;148:2845-2852. [ Links ]
24. Phipps RP, Stein SH, Roper RL. A new view of protaglandin E2 regulation of the immune response. Immunol Today 1992;12(10):349-352. [ Links ]
25. Van Der Pouw Kraan T, Boeije LCM, Smeenk RJT, Wijdenes J, Aarden LA. Prostaglandin E2 is a potent inhibitor of human Interleukin-12 production. J Exp Med 1995;181:775-779. [ Links ]
26. Bonta IL, Parnham MJ. Immunomodulatoryantiinflammatory functions of Etype prostaglandins. Minireview with emphasis on macrophage mediated effects. Int J Immunopharmac 1982;4(2):103-109. [ Links ]
27. Kunkel SL, Chensue SW, Phan SH. Prostaglandins as endogenous mediators of interleukin 1 production. J Immunol 1986;136(1):186-192. [ Links ]
28. Oppenheimer-Marks N, Kavanaugh A, Lipsky PE. Inhibition of transendothelial migration of human T lymphocytes by prostaglandin E2. J Immunol 1994;152:5703-5713. [ Links ]
29. Milano S, Arcoleo F, Dieli M, D'Agostino R, D'Agostino P, DeNucci G et al. Prostaglandin E2 regulates inducible nitric oxide synthase in the murine macrophage cell line J774. Prostaglandins 1995;49:105-115. [ Links ]
30. Kunkel SL, Wiggins RC, Chensue SW, Larrick J. Regulation of macrophage tumor necrosis factor production by prostaglandin E2. Biochem Biophys Res Commun 1986;137:404-410. [ Links ]
31. Boraschi D, Nierderhuber J. Regulation of macrophage suppression and cytotoxicity by interferon: Role of Ia-bearing macrophages. J Immunol 1982;129:1854-1858. [ Links ]
32. Alleva DG, Burger CJ, Elgert KD. Tumor growth increases 1a macrophage synthesis of tumor necrosis factor-a and prostaglandin E2: Changes in macrophage suppressor activity. J Leuk Biol 1993;53:550- 558. [ Links ]
33. Orino E, Sone S, Nii A, Ogura T. IL-4 up-regulates IL-1 receptor antagonist gene expression in human blood monocytes. J Immunol 1992;149:925-931. [ Links ]
34. Endo T, Ogushi F, Sone S. LPS-dependent cyclooxygenase-2 induction in human monocytes is down regulated by IL-13, but not by IFN-g. J Immunol 1996;156:2240-2246. [ Links ]
35. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Springgs MK et al. Human IL-17: A novel cytokine derived from T cells. J Immunol 1995;155:5483-5486. [ Links ]
36. Jovanovic DV, DiBattista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-1b and TNF-a, by human macrophages. J Immunol 1998; 160:3513-3521. [ Links ]
37. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med 1996;183:2593-2603. [ Links ]
38. Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1 induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol 1998;161:409-414. [ Links ]
39. Fournier T, Fadok V, Henson PM. Tumor necrosis factor-a inversely regulates prostaglandin D2 and prostaglandin E2 production in murine macrophages. J Biol Chem 1997;272:31065-31072. [ Links ]
40. Edwards III Hedegaard HB, Zlotnik A. Chronic infection due to Mycobacterium intracellulare in mice: Association with macrophage release of prostaglandin E2 and reversal by injection of indomethacin, muramyl dipeptide, or interferongamma. J Immunol 1986;136(5):1820-1827. [ Links ]
41. Stevens MG, Pugh Jr, GW, Tabatabai LB. Effects of Gamma interferon and indomethacin in preventing Brucella abortus infections in mice. Infect Immun 1992;60(10):4407-4409. [ Links ]
42. Filler SG, Ibe BO, Ibrahim AS, Ghannoum MA, Raj JU, Edwards Jr JE. Mechanisms by which Candida albicans induces endothelial cell prostaglandin synthesis. Infect Immun 1994;6(3):1064-1069. [ Links ]
43. Noverr MC, Phare SM, Toews GB, Coffey MJ, Huffnagle GB. Pathogenic yeast Cryptoccoccus neoformans and Candida albicans produce immunomodulatory prostaglandins. Infect Immun 2001;69(5):2957-2963. [ Links ]
44. Belley A, Chadee K. Eicosanoid production by parasites: From pathogenesis to immunomodulation? Parasitology Today 1995;11(9): 327-334. [ Links ]
45. Ramaswany K, Kumar P, H YX. A role for parasite-induced PGE2 in IL-10-mediated host immunoregulation by skin stage schistosomula of Schistosoma mansoni. J Immunol 2000;165:4567-4574. [ Links ]
46. Pinge-Filho P, Tadokoro CE, Abrahamsohn IA. Prostaglandins mediate suppression of lymphocyte proliferation and cytokine sintesis in acute Trypanosoma cruzi infection. Cell Immunol 1999;193:90-98. [ Links ]
47. DeFreitas LA, Mbow LM, Estay M, Bleyenberg JA, Titus RG. Indomethacin treatment slows disease progression and enhances a Th1 response in susceptible BALB/c mice infected with Leishmania major. Parasite Immunol 1999;21(5):273-277. [ Links ]
48. Titus RG, Sherry B, Cerami A. The involvement of TNF, IL1 and IL6 in the immune response to protozoan parasites. Immunol Today 1991; 12(3):A13-A16. [ Links ]
49. Pruzanski W, Vadas P. Phospholipase A2 a mediator between proximal and distal effectors of inflammation. Immunology Today 1991; 12(5):143-146. [ Links ]
50. Quiroga J, Prieto J. Liver cytoprotection by prostaglandins. Pharmac Ther 1993;58:67-92. [ Links ]
51. Yu Y, Chadee K. Entamoeba histolytica stimulates interleukin 8 from human colonic epithelial cells without parasite-enterocyte contact. Gastroenterology 1997;112:1536-1547. [ Links ]
52. Seydel KB, Li E, Swanson PE, Stanley Jr SL. Human intestinal epithelial cells produce proinflammatory cytokines in response to infection in a SCID mouse-human intestinal xenograft model of amebiasis. Infect Immun 1997;65:1631-1639. [ Links ]
53. Eckmann L, Reed SL, Smith JR, Kagnoff MF. Entamoeba histolytica trophozoites induce an inflammatory cytokine response by cultured human cells through the paracrine action of cytolytically released interleukin-1a. J Clin Invest 1995;96:1269-1279. [ Links ]
54. McGowan K, Piver G, Stoff JS, Donowitz M. Role of prostaglandins and calcium in the effects of Entamoeba histolytica on colonic electrolyte transport. Gastroenterology 1990;98:873-880. [ Links ]
55. Petri WA, Ravdin JI. Protection of gerbils from amebic liver abscess by immunization with the galactosespecific adherence lectin of Entamoeba histolytica. Infect Immun 1991;59:97-101. [ Links ]
56. Shibayama-Salas M, Tsutsumi V, MartínezPalomo A. Early invasive intestinal amebiasis in Mongolian gerbils. Arch Med Res 1992;23(2): 187-190. [ Links ]
57. Tsutsumi V, CanalesTreviño L, MartínezPalomo A. Immunoprophylaxis of experimental amebic liver abscess. Proc VIII Int Congress Protozoology; 1989 julio 10-17; Tsukuba, Japón. [ Links ]
58. Schain DC, Salata RA, Ravdin JI. Development of amebicidal cell mediated immunity in gerbils (Meriones unguiculatus) immunized with the galactoseinhibitable adherence lectin of Entamoeba histolytica. J Parasitol 1995;81(4):563-568. [ Links ]
59. Martínez-Palomo A, Kretschmer R, Meza I. Entamoeba histolytica and amebiasis. En: Ravdin JI, ed. Amebiasis. Human infection by Entamoeba histolytica. Nueva York:Wiley, 1988:143156. [ Links ]
60. Ortiz-Ortiz L, Zamacona G, Sepúlveda B, Capin NR. Cell mediated immunity in patients with amoebic abscess of the liver. Clin Immun Immunopath 1975;4:127-134. [ Links ]
61. Kretschmer RR, López-Osuna M. Mecanismos efectores e inmunidad antiamibiana. En: Krestchmer RR, ed. Amebiasis, infección y enfermedad por Entamoeba histolytica. México, D.F.: Editorial Trillas, 1994;135-149. [ Links ]
62. Ortiz-Ortiz L, Garmilla C, Tanimoto-Weki M, Zamacona-Ravelo G. Cellular hypersensivity in amebiasis. I. Reactions in hamsters inoculated with E. histolytica. Arch Invest Med 1973; Supl 1:141-146. [ Links ]
63. Salata RA, Ravdin JI. Review of the human immune mechanisms directed against Entamoeba histolytica. Rev Infect Dis 1986;8(2):261-269. [ Links ]
64. Gharidian E, Meerovitch E. In vitro amoebicidal activity of immune cells. Infect Immun 1982;36:243-246. [ Links ]
65. Salata RA, Martínez-Palomo A, Canales L, Murray HW, Treviño N, Ravdin JI. Suppression of T-lymphocyte responses to Entamoeba histolytica antigen by immune sera. Infect Immun 1990;58:3941-3946. [ Links ]
66. Stern JJ, Graybill JR, Drutz DJ. Murine amebiasis: The role of the macrophages in host defense. Am J Trop Med Hyg 1984;33(3):372-380. [ Links ]
67. Gharidian E, Meerovitch E. Macrophage requirement for host defense against experimental hepatic amebiasis in the hamster. Parasite Immunol 1982;4:219-225. [ Links ]
68. Denis M, Chadee K. In vitro and in vivo studies of macrophage functions in amebiasis. Infect Immun 1988;56:3126-3131. [ Links ]
69. Wang W, Chadee K. Entamoeba histolytica alters arachidonic acid metabolism in macrophages in vitro and in vivo. Immunology 1992;76: 242-250. [ Links ]
70. Wang W, Chadee K. Modulation of tumor necrosis factor production by macrophages in Entamoeba histolytica infection. Infect Immunol 1992;60(8):3169-3174. [ Links ]
71. Wang W, Chadee K. Entamoeba histolytica suppresses gamma interferon-induced macrophage class II major histocompatibility complex Ia molecule and IAb mRNA expression by a prostaglandin E2 dependent mechanism. Infect Immun 1995;63(3):1089-1094. [ Links ]
72. Seguin R, Keller K, Chadee K. Entamoeba histolytica stimulates the unstable transcription of c-fos and tumor necrosis factor-a mRNA by protein kinase C signal transduction in macrophages. Immunology 1995;86:49-57. [ Links ]
73. Sánchez-Ramírez B, Escalante B, Rosales-Encina JL, Talamás-Rohana P. Role of prostaglandin E2 on amoebic liver abscess formation in hamsters. Prostaglandins 1997;53:411-421. [ Links ]
74. Sánchez-Ramírez B, Escalante B, Rosales-Encina JL, Talamás-Rohana P. Entamoeba histolytica induces cyclooxygenase-2 expression in macrophages isolated from liver abscess. En: Tada I, Kojima S, Tsuji M, ed. Proc IX International Congress of Parasitolology; 1998 agosto 24-28; Makuhari Chiba, Japón. [ Links ]
75. Belley A, Keller K, Chadee K. Demonstration of cyclooxygenase-1 enzyme in Entamoeba histolytica: parasite production of Prostaglandin E2. 44Th Ann Meet Am Soc Trop Med Hyg; 1995 noviembre 17-21; San Antonio, Texas, USA. [ Links ]
76. Balsinde J, Balboa MA, Dennis EA. Functional coupling between secretory phospholipase A2 and cyclooxygenase-2 and its regulation by cytosolic group IV phospholipase A2. Proc Natl Acad Sci USA 1998;295(14): 7951-7956. [ Links ]
77. Ravdin JI, Murphy CF, Guerrant RL, Long-Krug SA. Effect of antagonists of calcium and phospholipase A on the cytopathogenicity of Entamoeba histolytica. J Infect Dis 1985;152:542-549. [ Links ]
78. Long-Krug SA, Fischer KJ, Hysmith RM, Ravdin JI. Phospholipase A enzymes of Entamoeba histolytica: Description and subcellular localization. J Infect Dis 1985;152:536-541. [ Links ]
79. Wang W, Keller K, Chadee K. Entamoeba histolytica modulates the nitric oxide synthase gene and nitric oxide production by macrophages for cytotoxicity against amoebae and tumour cells. Immunology 1994;83: 601-610. [ Links ]
80. Lin JY, Seguin R, Keller K, Chadee K. Transforming growth factor b1 primes macrophages for enhanced expression of the nitric oxide synthase gene for nitric oxide-dependent cytotoxicity against Entamoeba histolytica. Immunology 1995;85:400-407. [ Links ]
81. Barral-Netto M, Barral A, Brownell CE, Skeiky YA, Ellingsworth LR, Twardzik DR et al. Transforming growth factor-beta in leishmanial infection: A parasite escape mechanism. Science 1992;257:545-548. [ Links ]
(1) Facultad de Ciencias Químicas, Universidad Autónoma de Chihuahua, Chihuahua, México.
(2) Departamento de Patología Experimental, Centro de Investigación y de Estudios Avanzados-Instituto Politécnico Nacional. México, D.F., México.
Fecha de recibido: 15 de junio de 2001 · Fecha de aprobado: 17 de noviembre de 2001
Solicitud de sobretiros: Dra. Patricia Talamás-Rohana, Centro de Investigación y de Estudios Avanzados (Cinvestav)-Instituto Politécnico Nacional (IPN). Departamento de Patología Experimental, Avenida Instituto Politécnico Nacional # 2508, colonia San Pedro Zacatenco 07360 México, D.F., México.
Correo electrónico: ptr@mail.cinvestav.mx

