










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalud Pública de México
versión impresa ISSN 0036-3634
Salud pública Méx vol.43 no.4 Cuernavaca jul./ago. 2001
Factor de crecimiento transformante beta-1: estructura, función y mecanismos de regulación en cáncer
Oscar Peralta-Zaragoza, M. en C.,(1) A. Lagunas-Martínez, Q.B.P.,(1) Vicente Madrid-Marina, M. en C., Ph.D.(1)
Resumen
El factor de crecimiento transformante beta-1 (TGF-b1) es sintetizado por muchas estirpes celulares como linfocitos, macrófagos y células dendríticas, y su expresión regula de manera autócrina o parácrina la diferenciación, proliferación y el estado de activación de éstas y muchas otras células. En general, el TGF-b1 tiene propiedades pleiotrópicas en el contexto de la respuesta inmune durante el desarrollo de infecciones y procesos neoplásicos; sin embargo, los mecanismos de acción y regulación de la expresión de esta citocina aún no se comprenden del todo. En la presente revisión se describen las propiedades biológicas y los procesos moleculares que regulan la expresión del TGF-b1, para entender los efectos de esta citocina durante la proliferación y la diferenciación celular. El conocimiento de los mecanismos moleculares de la regulación del TGF-b1 puede representar una importante estrategia de tratamiento del cáncer. El texto completo en inglés de este artículo está disponible en: http://www.insp.mx/salud/index.html
Palabras clave: células neoplásicas; ciclo celular; Smad; factor beta tranformante de crecimiento
Abstract
Transforming growth factor beta-1 (TGF-b1) is produced by several cell lineages such as lymphocytes, macrophages, and dendritic cells, and its expression serves in both autocrine and paracrine modes to control the differentiation, proliferation, and state of activation of these and other cells. In general, TGF-b1 has pleiotropic properties on the immune response during the development of infection diseases and cancer; however, the mechanisms of action and regulation of gene expression of this cytokine are poorly understood, In this review, the biological properties and the molecular mechanisms that regulate TGF-b1 gene expression are described, to understand the role of this cytokine in growth and cell differentiation. The knowledge of molecular mechanisms of gene expression of TGF-b1 may serve to develop new cancer therapies. The English version of this paper is available at: http://www.insp.mx/salud/index.html
Key words: neoplasm cells; cell cycle; Smad; transforming growth factor beta
El factor de crecimiento transformante beta-1 (TGF-b1) es un regulador general de las actividades celulares con múltiples efectos biológicos, cuya identificación ha permitido entender los mecanismos por los cuales las funciones celulares están reguladas en la salud y alteradas en la enfermedad. El TGF-b1 fue identificado como un producto de células transformadas por el virus de sarcoma murino;1 sin embargo, actualmente se sabe que es sintetizado por diferentes tipos celulares incluyendo linfocitos, macrófagos, fibroblastos, miocitos, condrocitos, astrocitos, células epiteliales, células de riñón, células de placenta y plaquetas, así como por algunas células tumorales.2-4 A esta citocina se le denominó como el TGF-b por su función de inducir reversiblemente la transformación e inhibir la proliferación de fibroblastos normales, pero actualmente ha sido posible aislarlo de fuentes como el glioblastoma humano y de médula ósea bovina.
Los miembros de la super familia del TGF-b son reguladores multifuncionales de la proliferación y diferenciación de una amplia variedad de tipos celulares.5 Las isoformas TGF-b1, 2 y 3 comparten una alta homología de secuencia de aminoácidos (aa), tienen distintos patrones de expresión y sus funciones son mediadas por los mismos receptores5,6. El TGF-b1 es el miembro de la familia más ampliamente estudiado y puede actuar como un mitógeno indirecto sobre células mesenquimales o como estimulador de la síntesis de proteínas de matriz extracelular. Sin embargo, es también un potente inhibidor de la proliferación de células epiteliales, endoteliales, linfoides y mieloides.6 Resulta interesante que su efecto inhibidor in vitro esté ausente en muchas líneas celulares neoplásicas transformadas, incluyendo células de carcinoma epidermoide,7 de cáncer de mama8 y de carcinoma de pulmón,9 donde están alterados los mecanismos de regulación celular inducidos por el TGF-b1.
Isoformas y receptores del TGF-b
Existen varias isoformas del TGF-b designadas como TGF-b1, TGF-b2, TGF-b3, TGF-b4 y TGF-b5. Adicionalmente, un heterodímero del TGF-b1.2, se ha identificado en plaquetas porcinas. Las isoformas 1, 2, y 3 se encuentran codificadas en los cromosomas humanos 19q13, 1q41 y 14q24,10,11 mientras que las isoformas 4 y 5 se han identificado en aves y en anfibios, respectivamente. El TGF-b1 y el TGF-b2 son sintetizados por muchos tipos celulares, mientras que el TGF-b3 es expresado por células mesenquimales.10 Existe una homología de 70% de aminoácidos entre los TGF-b1 y TGF-b2, y de 79% entre los TGF-b2 y TGF-b3. Las cinco isoformas del TGF-b son sintetizadas como proteínas precursoras inactivas y contienen nueve residuos de cisteínas en el extremo C- terminal.12
Se han identificado cinco diferentes tipos de receptores (r) para el TGF-b que incluyen:
1) Los receptores funcionales, como son el tipo I (TbRI de 53 a 65 kilodaltones-kDa-), y el tipo II (TbRII de 83 a 110 kDa).
2) Los receptores no funcionales que incluyen el tipo III (TbRIII de 250 a 310 kDa), el tipo IV TbRIV de 60 kDa) y el tipo V (RV de 400 kDa).13
Los receptores TbRI, TbRII, TbRIII, y TbRV son co-expresados en la mayoría de las células analizadas, con excepción de pocas líneas tumorales. El TbRIV se ha identificado en células pituitarias.12 Se ha demostrado que existe una correlación entre la ausencia de los receptores TbRI y TbRII, y la pérdida de las respuestas celulares inducidas por el TGF-b1. Existe una afinidad diferencial entre TGF-b1, TGF-b2 y TGF-b3, y la unión por los diferentes receptores. Por ejemplo, TbRI y TbRII se unen al TGF-b1 y al TGF-b3 con mayor afinidad que al TGF-b2. Sin embargo, no hay relación directa entre la afinidad de unión y el efecto biológico, ya que el TGF-b2 es equivalente al TGF-b1 en la mayoría de las actividades biológicas. Los efectos de las isoformas del TGF-b están asociados a su disponibilidad, a la combinación de los tipos de receptores y a la vía de señalización intracelular que inducen.14,15
Todas las células normales y la mayoría de las células neoplásicas tienen receptores en su superficie para el TGF-b1. Los receptores RI y RII son los responsables de los efectos biológicos de TGF-b1 en las células de mamíferos; sin embargo, los receptores RIII, constituidas por los betaglicanos y la endoglina, son también capaces de unirse a TGF-b1.11 Los betaglicanos están ampliamente distribuidos en células mesenquimales, epiteliales y neuronas; se unen a través de su región extracelular de 100 kDa, poseen una región citoplásmica corta y una región intracelular que no participa en la transducción de señal. Se ha sugerido que el receptor TbRIII controla la disponibilidad del TGF-b1 en el microambiente extracelular local y regula su presentación activa a los receptores funcionales TbRI y TbRII. La endoglina es similar a los betaglicanos, particularmente en la región citoplásmica, y está presente en altos niveles en las células endoteliales. Los receptores TbRI, TbRII y betaglicanos se unen a los TGF-b1, 2 y 3, mientras que la endoglina se une a los TGF-b1 y 3.11
Funciones del TGF-b1
El TGF-b1 es miembro de una familia de polipéptidos que comparten características estructurales y funcionales. Es secretado en forma inactiva (latente), pero sus efectos biológicos y su unión a los receptores los realiza la forma activa. Su forma latente es un complejo formado por la unión del péptido maduro y el precursor del TGF-b1 (péptido asociado a latencia LAP),10 por lo que es sintetizado como una molécula pre-pro-TGF-b1 de aproximadamente 390 aa, con una secuencia señal de 29 aa, la cual es liberada para producir pro-TGF-b1. Varios sitios de glicosilación han sido identificados en la región N-terminal de pro-TGF-b1. La forma madura del TGF-b1 comprende una secuencia de 112 a 114 aa de la región C-terminal, la cual es liberada de pro-TGF-b1 por rompimiento en un sitio dibásico en el aa 278.10 La forma activa del TGF-b1 está constituida por dímeros de 25 kDa, que en condiciones de reducción generan monómeros de 12.5 kDa. Entre los mamíferos, la secuencia de aa del TGF-b1 es altamente conservada (100%), ya que es idéntica en humanos, cerdos, vacas y monos, y difiere sólo en 1 aa en los ratones.10 La estructura tridimensional de la proteína del TGF-b1 comienza en el extremo N-terminal con una cadena a-hélice (a1), seguida por una cadena b-plegada (b1) y una cadena b-plegada antiparalela irregular. A continuación sigue una segunda cadena a-hélice (a2) y un asa larga con numerosos contactos hidrofóbicos. Se continúa con una segunda cadena b-plegada (b2), otra asa larga y una tercera cadena a-hélice (a3), la cual termina con un giro b tipo II y un asa larga. El extremo C-terminal de la molécula forma una estructura b antiparalela extensa con un giro b tipo II. Las cadenas b3, b4, b5, b6 y b7 plegadas se forman por apareamiento de los residuos intercatenarios del extremo C-terminal de la proteína.16
La comparación de las estructuras del TGF-b1 y del TGF-b2 muestran mucha similitud conformacional; sin embargo, hay diferencias notables en la estructura local y en la flexibilidad, lo cual puede estar relacionado con diferencias en la unión al receptor y en las señales de transducción inducidas. Una diferencia estructural está localizada entre los residuos 69 a 72, donde la a-hélice más larga se une al extremo C-terminal de la proteína. Estos residuos forman un giro b tipo II en el TGF-b1, o bien, un giro b tipo I en el TGF-b2, lo cual puede tener un significado funcional, ya que esta sección de la molécula ha sido implicada en la unión al receptor. Los residuos 92 a 95 están implicados en el reconocimiento del dominio extracelular del receptor TbRII. Estos residuos forman un giro expuesto a la superficie tanto en el TGF-b1 y TGF-b2; sin embargo, en el TGF-b1 el giro es b tipo II y bien definido, mientras que en el TGF-b2 el giro está pobremente definido. Esta otra diferencia conformacional puede contribuir a una mayor afinidad del TGF-b1 por TbRII que por el TGF-b2.16
El TGF-b1 puede ser considerado como el prototipo de una citocina multifuncional, debido a los efectos que tiene sobre los diferentes blancos celulares. Es el inhibidor más potente de la proliferación de células de origen mieloide, mesenquimal, epitelial, linfoide, endotelial y de varios tipos de células malignas. Sin embargo, puede estimular la proliferación de fibroblastos normales, de células no epiteliales y ciertos tipos de células mesenquimales.6 Es un fuerte estimulador de la síntesis y depósito de proteínas de matriz extracelular (MEC) por parte de fibroblastos, osteoblastos y células endoteliales, además de aumentar la expresión de integrinas y receptores que median las interacciones celulares con proteínas de MEC.10 Streuli y colaboradores,17 han demostrado que la expresión del gen de TGF-b1 es afectada por la presencia o ausencia de proteínas de MEC, lo que sugiere una posible regulación negativa in vivo17. La disminución de las interacciones celulares con proteínas de MEC, induce la expresión del TGF-b1, lo cual conlleva a la síntesis de varias proteínas de MEC y sugiere un mecanismo de autorregulación.17
El TGF-b1 también induce otros eventos intracelulares como la regulación de factores de crecimiento que intervienen en la diferenciación celular; induce cambios de expresión de los genes jun-B, c-fos y c-myc; induce recambio de IP3; evita la fosforilación de pRb dependiente del contacto célula-célula, e induce la activación de proteínas G. En el contexto de la respuesta inmune, dependiendo de la concentración del TGF-b1, induce o inhibe la producción de citocinas por monocitos, pero inhibe la respuesta protectora de estas mismas células; promueve la quimiotaxis de fibroblastos, monocitos y neutrófilos; in vivo, aumenta las funciones efectora y de memoria de los linfocitos T CD4+ antígeno-específicos; estimula la secreción de IgA; inhibe la proliferación de linfocitos, células endoteliales, hepatocitos, queratinocitos y ciertas líneas celulares tumorales; inhibe la secreción de IgG e IgM; suprime la hematopoiésis dependiente de IL-3, la megacariopoiésis y la esteriodogénesis en las células adrenocorticales, e inhibe la diferenciación de adipocitos y miocitos.18-23 Además, se ha demostrado en estudios clínicos que puede inducir inmunosupresión en el transplante de órganos e injertos de piel, en el contexto de la activación de subpoblaciones de linfocitos T.24 En resumen, el TGF-b1 tiene un espectro muy amplio de funciones, las cuales dependen del estado de activación celular, de su concentración, del balance de expresión de otras citocinas y de las condiciones fisiológicas de las células sobre las que actúa; por este motivo, la distribución celular de su expresión in situ e in vivo está directamente asociada con el estado de diferenciación celular.
Ciclo celular y TGF-b1
Uno de los efectos más evidentes de la acción del TGF-b1 corresponde a la inhibición del ciclo celular. Su control selectivo de la expresión de genes puede ser indicativo de un mecanismo de inhibición de la proliferación por TGF-b1.4 Los puntos de control del ciclo celular entre las fases G1 a S y G2 a M, son los que activan los principales mecanismos de regulación del ciclo. Hay evidencias que sugieren que el TGF-b1 puede bloquear el ciclo celular en la transición de la fase G1 a la fase S, mediante la inducción de un conjunto de genes conocidos como inhibidores de cinasas dependientes de ciclinas (Kip).25 Los genes que codifican para los Kip están directamente involucrados en la regulación del ciclo celular, ya que inhiben la formación de los complejos activos de las cinasas dependientes de ciclinas (cdk), las cuales son las principales protagonistas en los puntos de control del ciclo celular.26 Dependiendo del tipo celular, el TGF-b1 in vitro induce la expresión de los genes Kip p15INK4B, p16, p21waf1/Cip1 y p27kip1; asimismo, inhibe la expresión de los genes c-myc, ciclina A, ciclina D1, ciclina E, cdk2, cdk4 y cdc25A.27 Además, el tratamiento con TGF-b1 de la línea celular HaCaT (células sensibles al TGF-b1) causa una rápida inducción de los genes p15 y p21, los cuales inhiben la actividad enzimática de las moléculas responsables de la fosforilación de pRB, cdk4/6-ciclina D y cdk2-ciclina E. Sin embargo, in vivo, en un modelo murino que carece del gene del TGF-b1 con genotipo heterocigoto (TGF-b1 +/-), la expresión de las proteínas y del RNAm de p15 y p21 tienen un patrón de regulación diferente notificado en estudios previos in vitro.28 Estos hallazgos son relevantes para definir las consecuencias funcionales del patrón de expresión de los genes inducidos por el TGF-b1, al igual que los efectos sobre la dinámica del ciclo celular.
Además, se ha propuesto que la inhibición de la proliferación de queratinocitos inducida por el TGF-b1 es una consecuencia de la inhibición de la transcripción del gen c-myc regulado por pRb.29 Las oncoproteínas virales E7 de virus de papiloma humano (HPV) E1A de adenovirus y antígeno T largo de SV40, son capaces de unirse al pRb e inhibir la expresión de TGF-b1. La participación del pRb en la inhibición de la transcripción de c-myc por el TGF-b1 sugiere que esta citocina puede actuar a través del pRb para inhibir a c-myc y reprimir la proliferación celular. Esta hipótesis predice que las mutaciones o deleciones del gen pRb podrían ser mecanismos por los cuales las células neoplásicas pueden superar los efectos de inhibición del ciclo celular inducidos por el TGF-b1.29
Por otra parte, el TGF-b1 puede inhibir la degradación de fosfatidilcolina por la activación de fosfolipasa C (PLC), lo cual es consistente con la idea de que la inhibición de la vía de degradación de fosfatidilcolina es un paso crítico para la inhibición de la proliferación celular por el TGF-b1.30 Sin embargo, no ha sido posible determinar si inhibe la degradación de fosfatidilcolina por su unión directa con PLC, o por alguna señal que influya sobre la secuencia de eventos de activación de PLC para la degradación de fosfatidilcolina. En conjunto, estas evidencias ayudan a comprender los mecanismos por los cuales el TGF-b1 puede regular de manera diferencial la proliferación celular de los distintos tipos celulares.
Transducción de señal inducida por TGF- b1
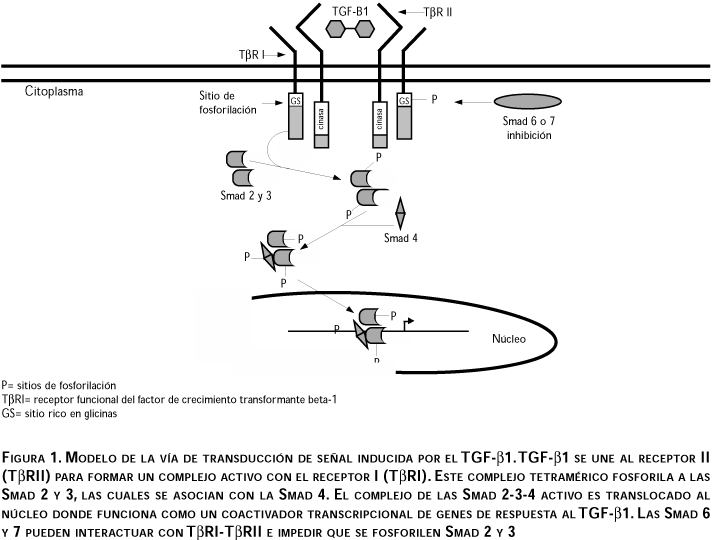
El TGF-b1 transduce señales por la unión a los receptores serina-treonina cinasa TbRI y TbRII; sin embargo, los efectos biológicos son mediados principalmente por RI31 (figura 1). Ambos receptores, TbRI y TbRII, tienen una región extracelular rica en cisteínas, una región transmembranal y una región citoplásmica, la cual contiene el dominio de serina-treonina cinasa. Los dominios de cinasa tienen 41% de similitud en su secuencia de aa.32 La región extracelular de TbRI es más corta que en TbRII. El TbRI tiene una secuencia GS (rica en glicina/serina) altamente conservada en la región que precede al dominio de cinasa próximo al N-terminal, la cual está ausente en TbRII.32 Inicialmente, al unirse el ligando (TGF-b1) a los receptores, TbRII se une al TGF-b1 directamente, mientras que TbRI sólo puede unirse al TGF-b1 cuando se encuentra coexpresado TbRII, presumiblemente como resultado de una cooperación funcional entre ambos.33 La expresión constitutiva de receptores produce la activación de TbRII, el cual puede homodimerizarse de manera independiente del ligando, a través de las regiones extracelular-transmembranal. Sin embargo, la homodimerización de TbRII no es capaz de transducir una señal funcional.33 En un modelo de activación de receptores para el TGF-b1 independiente del ligando, se ha demostrado que las células que carecen de TbRI requieren del acoplamiento de proteínas G para la transducción de señales inducidas por el TGF-b1.33
Respecto a la cooperación entre TbRI y TbRII, existe una dependencia funcional entre ambos. TbRI requiere de TbRII para unirse al ligando, mientras que TbRII requiere de TbRI para la señalización funcional.32,33 En presencia del TGF-b1, TbRI y TbRII tienen la habilidad para interactuar uno con otro y formar un complejo tetramérico a través del cual se transducen las señales. Este complejo molecular es la unidad receptora de señales que media las repuestas biológicas del TGF-b1.32,33 En este complejo, la región citoplásmica de TbRII está constitutivamente fosforilada, y fosforila el dominio GS de TbRI, lo cual induce su actividad cinasa. Las regiones intracelulares de ambos receptores tienen funciones distintas y ambas son necesarias para la transducción de señales.32,33 Las regiones transmembranal y extracelular son requeridas por TbRI y TbRII para la activación dependiente del ligando; sin embargo, se ha demostrado que la sobrexpresión de ambos receptores resulta en una combinación heteromérica de sus regiones citoplásmicas, produciendo una respuesta independiente del ligando.33 Además, la coexpresión de las regiones citoplásmicas de TbRI y TbRII, o la unión de TbRII con la región citoplásmica de TbRI, activa la respuesta celular de una manera independiente del TGF-b1. Estas evidencias sugieren que las regiones citoplásmicas de TbRI y TbRII interactúan física y funcionalmente en la formación de un complejo heteromérico.33 Por lo tanto, la unión del TGF-b1 estabiliza el complejo y genera respuestas fisiológicas adecuadas en una forma dependiente del ligando. La expresión anormal de TbRI y TbRII puede contribuir a la formación de complejos activos, produciendo una señalización independiente del ligando y, en consecuencia, respuestas celulares inapropiadas.33
Se ha notificado la participación de las proteínas transductoras de señales de la familia Smad* en las vías de transducción inducidas por el TGF-b.34-37 Estas proteínas tienen funciones inhibidoras y activadoras que median los efectos biológicos del TGF-b1. Las Smad 2, 3 y 4 son activadas después de la inducción de los receptores TbRI y TbRII. Cuando los receptores son estimulados por la unión del ligando, las Smad 2 y 3 son directamente fosforiladas por TbRI en un sitio consenso SS(V/M)S en el extremo C-terminal. Una vez fosforiladas las Smad 2 y 3, pueden formar un complejo estable con la 4. La proteína Smad 4 funciona como un punto de convergencia para las vías de señalización inducidas por los diferentes miembros de la superfamilia del TGF-b. El complejo activo formado por las Smads 2-3-4 es translocado al núcleo donde funciona como coactivador transcripcional y regula la transcripción de muchos genes que responden al TGF-b1 (figura 1).34 Las Smads inhibidoras (Smad 6 y 7) son inducidas por el TGF-b1, lo cual sugiere que hay un mecanismo de autorregulación negativa intracelular que regula la señal del TGF-b1.35 Las Smad 6 y 7 interactúan con TbRI en una manera más estable que las Smad 2 y 3 compitiendo por la unión con el receptor.35 Esto se puede deber a que las Smad 6 y 7 carecen del sitio de fosforilación en su extremo C-terminal. La sobre expresión de las Smad 6 o 7 inhibe la fosforilación de las Smad 2 y 3 estimulada por el ligando, lo cual sugiere que las Smad 6 y 7 tienen propiedades inhibidoras, ya que pueden bloquear la señalización en puntos tempranos de la vía de transducción de señal inducida por el TGF-b1. En general, la Smad 7 puede ser considerada como inhibidor de la señalización de la superfamilia del TGF-b37 (figura 1). Por otra parte, se han realizado interesantes avances en los mecanismos de la activación de receptores del TGF-b1 y las implicaciones de la regulación de los genes Mad (Mothers against dpp) en la transducción de señales por el TGF-b1, y se ha demostrado que estas proteínas son fosforiladas por TbRI.36 Esto fortalece la hipótesis de que el Mad puede interactuar con elementos reguladores específicos en genes blancos, e inducir la activación transcripcional de genes en respuesta a las señales transmitidas por el complejo TGF-b1/TbRII-TbRI.36
El promotor de TGF-b1
Para tener una mejor apreciación de los mecanismos de regulación de la expresión del TGF-b1, es imprescindible conocer este gen. El promotor de este gen humano fue identificado en 1989 por Kim y colaboradores,38 y a diferencia de otros promotores, posee características que lo hacen peculiar (figura 2). Se ha identificado la presencia de cuatro diferentes sitios de inicio de la transcripción, en las posiciones +1, +271, +470 y +525. Estos sitios de inicio son utilizados de manera diferencial dependiendo del estado de activación celular. Los dos primeros (+1, +271) son utilizados con mayor frecuencia que los otros dos (+470 y +525). El promotor no contiene las secuencias consenso TATA ni CAAT, y el porcentaje de secuencias GC es de 62%. Contiene 11 secuencias repetidas CCCCGCCCC, de las cuales siete corresponden a verdaderos sitios de unión para el factor transcripcional Sp1. Tiene tres secuencias homólogas al factor transcripcional FSE2, así como dos secuencias Erg1.38 En toda la secuencia de la región promotora se han identificado cinco regiones reguladoras, las cuales incluyen una región con actividad similar al enhancer, (secuencia de DNA con propiedades de potenciador) ubicada entre los nucleótidos -1132 a -731; dos regiones con actividad reguladora negativa, localizadas entre -1362 a -1132 y entre -731 a -453; y dos regiones con actividad de promotor basal que preceden a los dos primeros sitios de inicio de la transcripción. Figura 2. Además, se ha identificado una secuencia con actividad reguladora positiva entre los nucleótidos -453 a -323. Deleciones en esta secuencia inhiben la transcripción de la región distal del promotor.38 Respecto al segundo sitio de inicio de la transcripción (+271), se ha notificado que es muy activo, lo cual sugiere que existen secuencias localizadas después del primer sitio de inicio (+1), que son requeridas en la regulación de la expresión del TGF-b1. Como consecuencia, durante la transcripción del TGF-b1, se transcribe un RNAm a partir del segundo sitio de inicio del proceso, el cual parece ser regulado de manera independiente.38 La presencia de múltiples sitios de inicio de la transcripción puede ser común para genes que tienen un primer exón grande no traducido, los cuales pueden ser regulados de manera diferencial.
Dos regiones del promotor del TGF-b1 han sido identificadas como regiones de respuesta: una, a la autoinducción, y otra, a la inducción por ésteres de forbol (12-0 tetradecanoyl forbol-13-acetate, TPA).39-41 La primera región corresponde a la secuencia localizada entre los nucleótidos -454 a -323, y la segunda se encuentra entre los dos principales sitios de inicio de la transcripción (figura 2). Estas dos regiones contienen secuencias homólogas a elementos de respuesta a ésteres de forbol (TPA response element TRE), lo que sugiere que pueden actuar como reguladores inducibles activados por el tratamiento de las células con TPA.39-41 Se ha demostrado que los elementos TRE sirven como sitios de unión para el complejo AP-1. Además, la activación de proteína cinasa C por TPA resulta en un aumento de la expresión de los genes c-fos y c-jun, los cuales codifican para el factor transcripcional AP-1. En ambas regiones de respuesta del promotor del TGF-b1, la autoinducción es mediada por la unión de AP-1.39-41 La inducción de la expresión de c-jun por el TGF-b1, puede aumentar la expresión de este último durante el desarrollo normal y el proceso de oncogénesis.40 Se ha propuesto que la autoinducción del gen de TGF-b1 representa un mecanismo por el cual los efectos biológicos del TGF-b1 pueden ser amplificados, lo cual representa un mecanismo de regulación de la expresión de este gen.39-41
Se han identificado otros elementos reguladores en el promotor del gen del TGF-b1, entre los que se encuentran otras tres secuencias de unión a AP-1. Una de ellas, de alta afinidad, se localiza en la secuencia de -420 a -412, y las otras dos, de baja afinidad, están localizadas entre los nucleótidos de +155 a +170 y de +247 a +289.40 La identificación de un elemento TRE en el extremo 3' después del último exón, sugiere la presencia de un enhancer que puede tener una función importante en la regulación transcripcional del TGF-b1.40 Una secuencia similar a la secuencia consenso del elemento NF-1 se ha identificado en la posición de -260 a -240; sin embargo, no parece tener una función importante en la autoinducción del TGF-b1 (figura 2). 39 En conjunto, todas estas características del promotor del TGF-b1 sugieren la participación de complejos procesos moleculares de regulación de la expresión de este gen.
Regulación de la expresión génica del TGF-b1
Uno de los primeros niveles de regulación del TGF-b1 es el transcripcional, donde su expresión génica puede ser mediada por las interacciones de los factores transcripcionales con los diferentes elementos reguladores descritos en el promotor. En las cinco isoformas del TGF-b, la regulación de la transcripción es diferente e independiente, ya que cada una de ellas tiene sus propios promotores con diferentes elementos reguladores, que son selectivamente activados por señales específicas durante la activación o diferenciación celular.42 Existen evidencias que al parecer apoyan que el gen del TGF-b1 puede funcionar como blanco para una variedad de mecanismos reguladores, incluyendo los oncogenes. Se ha notificado la inducción de actividad promotora del TGF-b1 por los oncogenes jun, fos, ras y tax, al igual que por Rb de manera positiva o negativa, dependiendo del tipo celular, así como por el gen E1A de adenovirus.43 Además, se ha demostrado que el producto del gen E6 de HPV-16 tiene propiedades de transactivación del promotor del TGF-b1.44 Esta transactivación es una función específica de la proteína E6 inducida a través de un elemento rico en GC (guanina-citocina) localizado entre los nucleótidos -109 a -100. Esta secuencia rica en GC también corresponde al sitio de unión para el factor transcripcional Sp1. La dualidad de funciones para una secuencia de DNA no es rara, ya que puede funcionar como un elemento regulador para un factor transcripcional como Sp1, y al mismo tiempo representa un blanco de transactivación como lo es para E6 de HPV-16.44
Por otra parte, Scotto y colaboradores identificaron que el transcrito principal del TGF-b1 humano tiene 2 136 pb43, el cual es más corto que el originalmente descrito.45 La reducción en el tamaño del RNAm puede explicarse por el proceso de poliadenilación que ocurre en diferentes sitios, como por ejemplo en la secuencia ATTAAA de la posición +2 136 y no en la secuencia AATAAA de la posición +2 517.43 Además, varios estudios han documentado la presencia de transcritos del TGF-b1 de menor tamaño en células humanas. El hecho de que el promotor del gen del TGF-b1 contenga múltiples sitios de inicio de la transcripción, puede explicar la existencia de estos transcritos de menor longitud.43
La regulación postranscripcional representa otro nivel importante de control de la regulación de la expresión del TGF-b1.46 Se ha identificado que la región 5' no traducida (5'-UTR) del TGF-b1 tiene una función importante en la regulación postranscripcional. El RNAm correspondiente al 5'-UTR tiene un tamaño de 840 nucleótidos y es altamente rico en secuencias GC. Evidencias experimentales han demostrado que, al insertar la secuencia de +11 a +147 del 5´-UTR del TGF-b1 en el 5´-UTR del gen de la hormona de crecimiento, se inhibe la expresión del gen heterólogo de manera célula-específica.46 El análisis de la región 5´-UTR de TGF-b1 ha permitido identificar la presencia de una estructura secundaria tallo-asa estable que comprende del nucleótido +49 al +76, y se ha demostrado que esta estructura es suficiente para inhibir tanto la expresión del TGF-b1 como la expresión de genes heterólogos. Además, la secuencia de DNA del 5'-UTR puede contener sitios de unión para factores transcripcionales específicos para cada tipo celular, los cuales pueden regular la traducción del RNAm del TGF-b1.46 Sin embargo, se requieren más estudios para entender los mecanismos moleculares mediante los cuales las proteínas de unión al DNA pueden modular la expresión postranscripcional del TGF-b1. También se ha notificado que el control de la síntesis del TGF-b1 por esteroides y otras moléculas relacionadas, se efectúa a nivel postranscripcional.46
Los mecanismos y niveles de regulación de la expresión génica del TGF-b1 son complejos durante las diferentes respuestas celulares. Puede regular de manera positiva o negativa a muchos genes por medio de sus promotores. De igual manera, la expresión génica del TGF-b1 es regulada a través de su región promotora por varios factores transcripcionales. Esto sugiere que su gen tiene mecanismos complejos de regulación.42 Además, en las respuestas inducidas por él hay una expresión diferencial de genes que participan en la transducción de señal, en la regulación del ciclo celular y en la diferenciación celular. En muchos casos, esta expresión diferencial de genes es regulada de manera espacio-tiempo-célula específica por el TGF-b1, por lo que las respuestas celulares a su inducción comprenden la integración de los mecanismos de regulación génica y de diferenciación celular. En consecuencia, alteraciones en su patrón de expresión y en sus mecanismos de acción, se han relacionado con desórdenes de la respuesta inmune -que a su vez se asocian con el desarrollo de autoinmunidad y procesos neoplásicos-, susceptibilidad a infecciones oportunistas y fibrosis asociada con procesos inflamatorios crónicos.47
Función del TGF-b1 en la oncogénesis
Los TGF-b regulan la proliferación de muchas células normales; sin embargo, en las células tumorales ocurre la pérdida de respuesta a esta citocina. Varias evidencias indican una correlación entre el aumento de la expresión de la proteína y del RNAm del TGF-bs con los estados avanzados de la tumorigénesis, así como la disminución de la sobrevida en cánceres epiteliales, neuroectodérmicos y de origen mesenquimal.48-53 Estos hallazgos sostienen la hipótesis de que el aumento de la expresión del TGF-b1 lleva a la pérdida de la respuesta inhibidora de la proliferación celular inducida por éste, y puede representar un mecanismo de escape de la célula tumoral que favorece la evolución del proceso neoplásico.51 Se ha demostrado que el carcinoma de colon y el glioblastoma tienen una pérdida progresiva de la respuesta inhibidora de la proliferación por el TGF-b1, lo cual varía directamente con la evolución del tumor.54,55 Los mecanismos que aumentan la expresión del TGF-b1 pueden explicarse en parte por los procesos de regulación de la expresión de este gen; sin embargo, una vez que la célula tumoral pierde su capacidad para inhibir su proliferación por el TGF-b1, produce cantidades masivas de esta citocina. Esta condición proporciona una ventaja selectiva para la sobrevivencia de la célula tumoral, ya que el TGF-b1 induce de novo la angiogénesis y el desarrollo de un estado de inmunosupresión, incluyendo la inhibición de la respuesta antitumoral de las células NK.56,57 Además, el TGF-b1 tiene funciones importantes en la regulación del ciclo celular, por lo que no es sorprendente que las alteraciones genéticas asociadas con el desarrollo del cáncer ocurran en genes que están involucrados en la regulación de la proliferación celular por el TGF-b1. Defectos en los receptores del TGF-b1, en genes involucrados en la transducción de señal por el TGF-b1 y en proteínas que regulan el ciclo celular relacionadas con éste, tienen implicaciones importantes en el proceso oncogénico.51 A continuación se discutirá la importancia de las alteraciones de los receptores, así como de las vías de transducción de señal inducidas por el TGF-b1, en el desarrollo de diferentes tumores.
Alteraciones en los receptores de TGF-b en cáncer
En el contexto de la proliferación celular, la principal función de los receptores del TGF-b1 es transducir las señales para mantener a las células detenidas en la fase G1 del ciclo celular. Mutaciones en los genes involucrados en las vías de señalización por el TGF-b1, podrían permitir a la célula tumoral escapar a la inhibición mediada por él. Los receptores TbRI y TbRII para el TGF-b1 son requeridos para la transducción de señal, y la pérdida de cualquiera de ellos podría causar a la célula tumoral una incapacidad para responder al TGF-b1. La función de los receptores del TGF-b1 en la tumorigénesis es ejemplificada por mutaciones somáticas que inactivan a TbRII, asociadas con la pérdida de la inhibición de la proliferación por el TGF-b1. Estas mutaciones se han identificado en cáncer colorectal no poliposo hereditario (HNPCC, por sus siglas en inglés) y en carcinoma endometrial, asociadas con inestabilidad de microsatélites y referidas como errores de replicación y pérdida de heterocigocidad en los genes de reparación del DNA.58 Estas mutaciones de inactivación se han identificado en un alelo del gen de TbRII en 57% de los adenomas de pacientes con HNPCC, y se ha determinado la ausencia del segundo alelo en 85% de los cánceres colorectal es que han progresado a un estado invasivo.59 Se ha demostrado que el gen de TbRII tiene alteraciones genéticas, deleciones y pérdida del gen homocigoto en cánceres de cabeza y cuello, gástrico, de mama, en células pequeñas de carcinoma de pulmón y en carcinoma endometrial.58-61 En relación con las alteraciones genéticas en el gen de TbRI, se les han identificado en líneas celulares de carcinoma de próstata, las cuales causan ausencia de expresión de TbRI e insensibilidad para la inhibición de la proliferación por el TGF-b1. La respuesta al TGF-b1 es restaurada por la transfección de cDNA de TbRI. Además, la rexpresión de TbRII en líneas celulares de cáncer de mama, restaura la respuesta al TGF-b1 e inhibe la capacidad de estas células para inducir la formación de tumores en ratones desnudos.62 En consecuencia, tanto TbRI como TbRII tienen mutaciones o disminución de su expresión del gen o de la proteína en muchos cánceres humanos, por lo que es importante el estudio de los mecanismos de regulación de la expresión de los receptores TbRI y TbRII del TGF-b1 en el desarrollo de tumores.
Alteraciones de la transducción de señal del TGF-b1 en cáncer
En la vía de transducción de señal inducida por el TGF-b1, las proteínas Smad son un blanco con una alta frecuencia de mutaciones con potencial tumorigénico, las cuales transforman a las células tumorales para superar la inhibición de la proliferación inducida por el TGF-b1. Por ejemplo, el gen DPC4 (deleted in pancreatic cancer locus 4), el cual codifica para la Smad 4, adquiere su nombre por el alto porcentaje de mutaciones (90%) y deleciones homocigotas que ocurren en el cáncer pancreático.63 Se ha demostrado que el gen DPC4 puede ser inactivado por mutaciones somáticas en tumores de cabeza y cuello, mama, colon y adenocarcinomas de pulmón.64 La pérdida de este gen también se ha identificado en células tumorales que no responden a la inhibición de la proliferación por el TGF-b1, y la respuesta es restaurada por la transfección del gen DPC4 en estas mismas células.64 Los estudios de cristalografía de rayos X y el análisis mutacional de DPC4 han mostrado que la mayoría de las mutaciones residen en el dominio MH2 C-terminal, el cual interactúa con otras Smad.65 Mutaciones en este dominio de la Smad 4 se han identificado en cánceres de páncreas, colon, esófago, mama, ovario, cabeza y cuello,63-65 asimismo, mutaciones en el mismo dominio de la Smad 2 se han asociado con cánceres de colon, cabeza y cuello.65 Además, se ha identificado una mutación sin sentido en una arginina en el dominio MH1 de unión con el DNA de las Smad 2 y 4, así como otras mutaciones prevalentes en cánceres humanos.64,65 Esta mutación sin sentido aumenta la afinidad del dominio N-terminal por el dominio C-terminal de la Smad 2, evitando su interacción con la Smad 4 y generando un mecanismo autoinhibidor para la activación de esta última proteína. En conclusión, la presencia de mutaciones o la represión de la expresión de los genes TbRI y TbRII, o de algunos de los elementos de la vía de transducción de señal inducida por el TGF-b1, son de importancia capital en el desarrollo de muchos cánceres humanos. Los mecanismos de regulación de la expresión génica de estas moléculas se encuentran en estudio, por lo que el conocimiento de los eventos moleculares asociados con el escape de las células tumorales a la regulación por el TGF-b1, aportará más bases para entender los mecanismos de transformación oncogénica.
Conclusiones
La multifuncionalidad del TGF-b1, así como los mecanismos moleculares de regulación descritos en el presente artículo, no son las únicas propiedades que distinguen a esta citocina. Como otros autores, consideramos que la descripción que mejor define a esta citocina tan peculiar, es la realizada por Sporn MB,66 quien dice: "La función de TGF-b1 [...] no es tener una acción intrínseca, sino servir como un mecanismo para el acoplamiento de la célula a su entorno, de tal manera que la célula tiene la plasticidad para responder apropiadamente a los cambios en el medio ambiente o cambiar en su estado de diferenciación.6" Esto significa que la multifuncionalidad del TGF-b1 está acompañada por un aumento de su expresión por la activación de sus receptores, por la vía de transducción de señal que induce y por la regulación del ciclo celular; todo lo cual sirve para aumentar las respuestas o inhibir las actividades de las diferentes poblaciones celulares.
Un aspecto interesante para analizar es la propiedad que tiene el TGF-b1 para desarrollar mecanismos de evasión de la respuesta inmune por parte de las células cancerosas. Hay trabajos que evidencian claramente el escape de la vigilancia inmune del huésped durante el desarrollo del tumor, donde se ha demostrado que el TGF-b1 estimula la producción de interleucina IL-10 por parte de macrófagos en modelos murinos con timomas tanto in vitro como in vivo.67 Estas evidencias apoyan que los tumores son capaces de producir el TGF-b1 y, en consecuencia, inducir el desarrollo de mecanismos de escape de la vigilancia inmune. Por ejemplo, en ese mismo modelo se demostró un aumento en el perfil de citocinas tipo Th2 (T-cell helper 2) y una disminución del tipo Th1.67 Sin embargo, la adición de anticuerpos anti-IL-10 inhibe la producción de citocinas Th2 y estimula la respuesta a IL-2. Por lo tanto, adicional al efecto del TGF-b1, la sobreproducción de IL-10 inducida por el TGF-b1, puede representar un factor de supresión de la respuesta inmune en este tipo de tumores. Adicionalmente, en el laboratorio de la Dirección de Biología Molecular de Patógenos, del Instituto Nacional de Salud Pública, se ha demostrado que existe una estrecha relación entre el desarrollo del cáncer cervicouterino y la expresión del TGF-b1.** Estas evidencias son fuerte indicio de que el propio tumor es quien produce el TGF-b1, y probablemente, como se ha descrito en otros modelos estudiados, esto permita el desarrollo de un mecanismo de evasión de la respuesta inmune antitumoral.
Finalmente, el avance en el conocimiento de las funciones del TGF-b1, ha permitido entender no sólo los mecanismos de regulación de la expresión de esta citocina, sino también los mecanismos de inhibición de la proliferación celular, y comprender que las alteraciones en los receptores y/o vías de transducción de señal inducidos pueden contribuir al desarrollo del proceso neoplásico. Como se mencionó, se han identificado alteraciones del TGF-b1 y de sus receptores en muchos cánceres humanos. Sin embargo, antes de intentar restaurar estas condiciones anormales con medidas terapéuticas como el uso exógeno del TGF-b1 o de agentes que inducen su expresión, los tratamientos y programas de quimioprevención clínica deben estar fundamentados en los mecanismos de la carcinogénesis y deben determinar en qué nivel de la progresión maligna se encuentra alterada la maquinaria que regula la inhibición de la proliferación celular inducida por el TGF-b1. Muchos de estos conocimientos se han adquirido gracias al análisis funcional in vitro y de los modelos animales in vivo, incluyendo los de animales que carecen del gen (animales knock-out) y transgénicos del TGF-b1, los cuales ponen de manifiesto que las terapias clínicas basadas en la modulación de esta citocina representan una importante estrategia para el tratamiento de tumores.
Referencias
1. De Larco JE, Todoro GJ. Growth factors form murine sarcome virus-transformed cells. Proc Natl Acad Sci USA 1978;75:4001-4005. [ Links ]
2. Da Cunha A, Vitkovic L. Transforming growth factor-beta 1 (TGF-beta) expression and regulation in rat cortical astrocytes. J Neuroimmunol 1992; 36:157-169. [ Links ]
3. Steigerwalt RW, Rundhaug JE, Nettesheim P. Transformed rat tracheal epithelial cells exhibit alterations in transforming growth factor-beta secretion and responsiveness. Mol Carcin 1992;5:32-40. [ Links ]
4. Lyons RM, Moses HL. Transforming growth factor and the regulation of cell proliferation. Eur J Biochem 1990;187:467-523. [ Links ]
5. Massagué J. Transforming growth factor beta family. Annu Rev Cell Biol 1990;6:597-641. [ Links ]
6. Sporn MB, Roberts AB. Transforming growth factor-beta: Recent progress and new challenges. J Cell Biol 1992;119:1017-1021. [ Links ]
7. Shipley GD, Pittelkow MR, Wille JJ, Scott RE, Moses HL. Reversible inhibition of normal human prokeratinocyte proliferation by type beta transforming growth factor-growth inhibitor in serum-free medium. Cancer Res 1986;46:2068-2071. [ Links ]
8. Knabbe C, Lippman ME, Wakefield LM, Flanders FC, Kasid A, Derynck R et al. Evidence that transforming growth factor-beta is a hormonally regulated negative growth factor in human breast cancer cells. Cell 1987; 48:417-428. [ Links ]
9. Masui T, Wakefield LM, Lechner JF, LaVeck MA, Sporn MB. Type beta transforming growth factor is the primary differentiation inducing serum factor for normal human bronchial epithelial cells. Proc Natl Acad Sci USA 1986;83:2438-2442. [ Links ]
10. Barnard JA, Lyons RM, Moses HL. The cell biology of transforming growth factor b. Biochem Biophys Acta 1990;1032:79-87. [ Links ]
11. Cheifetz S, Hernández H, Laiho M, Ten-Dijke P, Iwata KK, Massagué J. Distinct transforming growth factor beta receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J Biol Chem 1990;265:20533-20538. [ Links ]
12. Massague J, Cheifetz S, Ignotz RA, Boyd FT. Multiple type-beta transforming growth factors and their receptors. J Cell Physiol Suppl. 1987; suppl 5:43-47. [ Links ]
13. Massague J. Receptor for the TGF-beta family. Cell 1992;69:1067-1070. [ Links ]
14. Wrana JL, Attisano L, Carmaso J, Zentella A, Doody J, Laiho M et al. TGF-beta signals through a heteromeric protein kinase receptor complex. Cell 1992;71:1003-1014. [ Links ]
15. Lin HY, Wang XF. Expression cloning of TGF-beta receptors. Mol Repord Dev 1992;32:105-110. [ Links ]
16. Hinck AP, Archer SJ, Qian SW, Roberts AB, Sporn MB, Weatherbee JA et al. Tranforming growth factor b1: Three-dimensional structure in solution and comparison with the X-ray structure of transforming growth factor b2. Biochem 1996 (26) Jul 2;35:8517-8534. [ Links ]
17. Streuli CH, Schmidhauser C, Kobrin M, Bissell MJ, Derynck R. Extracellular matrix regulates of the TGF-b1 gene. J Cell Biol 1993;120:253-259. [ Links ]
18. Palladino MA, Morris RE, Starnes HF, Levinson AD. The transforming growth factor-betas. A new family of immunoregulatory molecules. Ann NY Acad Sci 1990;593:181-187. [ Links ]
19. Okadome T, Yamashiya H, Franzen P, Moren A, Sheldin CH, Miyazono K. Distinct roles of the intracellular domains of transforming growth factor-b type I and type II receptors in signal transduction. J Biol Chem 1994; 269:30753-30756. [ Links ]
20. Ohtsuki M, Massagué J. Evidences for the involvement of protein kinase activity in transforming growth factor-beta signal transduction. Mol Cell Biol 1992;12:261-265. [ Links ]
21. Rizzino A. Transforming growth factor-b: Multiple effects on cell differentiation and extracellular matrices. Dev Biol 1988;130:411-422. [ Links ]
22. Kimberly H, Morris L, Braun L, Biron CA. A role for transforming growth factor-b1 in regulating natural killer cell and T lymphocyte proliferative responses during acute infection with lymphocytic choriomeningitis virus. J Immunol 1991;147:2717-2727. [ Links ]
23. Fontana A, Frei K, Bodmer S, Hofer E, Schreier MH, Palladino MA et al. Transforming growth factor-b inhibit the generation of cytotoxic T cell in virus infected mice. J Immunol 1989;143:3230-3234. [ Links ]
24. Sporn MB, Roberts AB. Transforming growth factor-beta. Multiple actions and potential clinical applications. SAMA 1989 Aug18;262(7): 938-941. [ Links ]
25. Polyak K, Kato JY, Solomon MJ et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994 Jan; 8(1): 9-22. [ Links ]
26. Peralta-Zaragoza O, Bahena-Román M, Madrid-Marina V. Regulación del ciclo celular y desarrollo del cáncer: perspectivas terapéuticas. Salud Publica Mex 1997;39(5):451-462. [ Links ]
27. Ravitz MJ, Wenner CE. Cyclin-dependent kinase regulation during G1 phase and cell cycle regulation by TGF-b1. Adv Cancer Res 1997;71,165-207. [ Links ]
28.Tang B, Bottinger PE, Jakowlew JB. Transforming growth factor b1 is a new form of tumor suppressor with true haploid inssufficiency. Nat Med 1998;4:802-807. [ Links ]
29.Pietenpol AJ, Stein WR, Moran E. TGF-b1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 1990;61:777-785. [ Links ]
30. Díaz-Meco MT, Domínguez I, Sanz L, Municio MM, Berra E, Cornet ME et al. Phospolipasa C-mediated hydrolisis of phosphatidylcholine is a target of transforming growth factor-b1 inhibitory signals. Mol Cell Biol 1992; 12:302-308. [ Links ]
31. Ohtsuki M, Massagué J. Evidences for the involvement of protein kinase activity in transforming growth factor-b1 signal transduction. Mol Cell Biol 1992;12:261-265. [ Links ]
32. Massagué J. TGFb signaling: Receptors, transducers and Mad proteins. Cell 1996;85:947-950. [ Links ]
33. Feng XH, Derynck R. Ligand-independent activation of transforming growth factor (TGF)-b1 receptors. J Biol Chem 1996;271:13123-13129. [ Links ]
34.Heldin HC, Miyazomo K, Dijke TP. TGF-b signalling from cell membrane to nucleus through SMAD proteins. Nature 1997;390:465-471. [ Links ]
35. Derynck R, Zhang Y, Feng HX. Smads: Transcriptional activators of TGF-b responses. Cell 1998;95:737-740. [ Links ]
36. Attisano L, Wrana LJ. Mads and Smads in TGF-b signalling. Curr Opin Cell Biol 1998;10:188-194. [ Links ]
37. Whitman M. Feedback from inhibitory SMADs. Nature 1997;389: 549-550. [ Links ]
38. Kim SJ, Glick AB, Sporn MB, Roberts AB. Characterization of the promoter region of the human transforming growth factor-b1 gene. J Biol Chem 1989;264:402-408. [ Links ]
39. Kim SJ, Jeang KT, Glick AB, Sporn MB, Roberts AB. Promoter sequences of the human transforming growth factor-b1 gene responsive to transforming growth factor-b1 autoinduction. J Biol Chem 1989;264:7041-7045. [ Links ]
40. Kim SJ, Angel P, Lafyantis R, Hattorri K, Kim YK, Sporn MB et al. Autoinduction of transforming growth factor-b1 is mediated by the AP-1 complex. Mol Cell Biol 1990;10:1492-1497. [ Links ]
41. Kim SJ, Denhez F, Kim KY, Holt JT, Sporn MB, Roberts AB. Activation of the second promoter of the transforming growth factor-b1 gene by transforming growth factor-b1 and phorbol ester occurs through the same sequences. J Biol Chem 1989;264:19373-19378. [ Links ]
42. Roberts AB, Kim SJ, kondaia HP, Jakowlew SB, Dunhez F, Glick AB et al. Transcrptional control of expression of the TGF-beta. Ann NY Acad Sci.1990;593:43-50. [ Links ]
43. Scotto L, Vaduva PI, Wager RE, Assoian RK. Type b1 transforming growth factor gene expression. J Biol Chem 1990;265:2203-2208 [ Links ]
44. Dey A, Atcha AI, Bagchi S. HPV-16 E6 oncoprotein stimulates the transforming growth factor-b1 promoter in fibroblasts through a specific GC-rich sequence. Virology. 1997;228:190-199. [ Links ]
45. Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK et al. Human transforming growth factor-b complementary DNA sequence and expression in normal and transformed cells. Nature 1985;316: 701-705. [ Links ]
46. Kim SJ, Park K, Koeller D, Kim KY, Wakefield LM, Sporn MB et al. Post-transcriptional regulation of the human transforming growth factor-b1 gene. J Biol Chem 1992;267:13702-13707. [ Links ]
47. Letterio JJ, Roberts BA. Regulation of immune responses by TGF-b1. Annu Rev Immunol 1998;16:137-161. [ Links ]
48. Jennings MT, Kaariainen BA, Gold LI, Maciunas RJ, Commers BA. TGF-b1 and TGF-b2 are potencial growth regulators for medulloblastomas, primitive neuroectodermal tumors and ependymomas: Evidence in sopport of an autocrine hypothesis. Hum Pathol 1994;25:464-475. [ Links ]
49. Gold L, Korc M. Expression of TGF-b1, 2 and 3 mRNA and protein in human cancers. Dig Surg 1994; 11:150-156. [ Links ]
50. Naef M, Ishiwata T, Friess H, Buchler M, Gold LI, Korc M. Differential localization of transforming growth factor-B isoforms in human gastric mucosa and over-expression in gastric carcinoma. Int J Cancer 1997;71: 131-137. [ Links ]
51. Filmus J, Kerbel RS. Development of resistance mechanisms to the growth inhibitory effects of transforming growth factor-b during tumor progression. Curr Opin Oncol 1993;5:123-129. [ Links ]
52. Friess H, Yamanka Y, Buchler M, Ebert M, Beger HG, Gold LI et al. Enhanced expression of transforming growth factor-b isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105:1846-1856. [ Links ]
53. Gold LI, Saxena B, Zagzag D, Miller DC, Koslow M, Brandeis L et al. Increased expression of TGF-b isoforms by malignant gliomas. J Cell Biol 1992;16B:122. [ Links ]
54. Hsu S, Huang F, Hafez M, Winawer S, Friedman E. Colon carcinoma cell swich their response to transforming growth factor-b1 with tumor progression. Cell Growth Differ 1994;5:267-275. [ Links ]
55. Jennings MT, Maciunas RJ, Carver R, Bascom CC, Juneau P, Misulis K et al. TGF-b1 and TGF-b2 are potencial growth regulators for low-grade and malignant gliomas in vitro: Evidence in support of an autocrine hypothesis. Int J Cancer 1991;49:129-139. [ Links ]
56. Yang EY, Moses HL. Transforming growth factor-b1 induced changes on cell migration, proliferation and angiogenesis in the chicken allantoic membrane. J Cell Biol 1990;111:731-741. [ Links ]
57. Wallick SC, Figari IS, Morris RE, Levinson AD, Palladino MA. Immunoregulatory role of transforming growth factor-b in development of killer cells: Comparison of active and latent TGF-b1. J Exp Med 1990;172: 1777-1784. [ Links ]
58. Myeroff LL, Parson R, Kim SJ, Hedrick L, Cho KR, Orth K et al. A transforming growth factor-b receptor type II gene mutation common in colon and gastric rare in endometrial cancer with microsatellite inestability. Cancer Res 1995; 55:5545-5547. [ Links ]
59. Akiyama Y, Iwanaga R, Seitoh K, Shiba K, Ushio K, Ikeda E et al. Transforming growth factor-b type receptor II gene mutations in adenomas from hereditary non-polyposis colorectal cancer. Gastroenterology 1997; 112:33-39. [ Links ]
60. Fynan TM, Reiss M. Resistance to inhibition of cell growth by transforming growth factor-b and its role in oncogenesis. Crit Rev Oncog 1993; 4:493-540. [ Links ]
61. Garrigue AL, Muñoz ASJ, Antonia SJ, Gesmonde J, Velucci VF, Reiss M. Missense mutations of transforming growth factor-b type II receptor in human head and neck squamous carcinoma cells. Cancer Res 1995;55: 3982-3987. [ Links ]
62. Sun L, Wu G, Willson JK, Zborowska E, Yang J, Rajkarunanayake Y et al. Expression of transforming growth factor-b type II receptor leads to reduced malignancy in human breast cancer MCF7 cells. J Biol Chem 1994; 269:26449-26455. [ Links ]
63. Hahn SA, Schutte M, Shamsul HATM, Moskaluk CA, Da Costa LT, Roxenblum E et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350-356. [ Links ]
64. Schutte M, Hruban RH, Hedric KL, Cho KR, Nadasdy GM, Weinstein CL et al. DPC4 gene in various tumor type. Cancer Res 1996 Jun 1;56(II): 2527-2530. [ Links ]
65. Hata A, Lo RS, Wotton D, Lagna G, Massague J. Mutations increasing autoinhibition inactive tumour suppressors Smad 2 and Smad 4. Nature 1997;388:82-87. [ Links ]
66. Sporn MB, Roberts AB. TGF-b: Problems and prospects. Cell Regul 1990;1:875-882. [ Links ]
67. Maeda H, Kuwahara H, Ichimura Y, Ohtsuki M, Kurakata S, Shiraishi A. TGF-beta enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J Immunol 1995 Nov 15;155(10)4926-4932. [ Links ]
Este trabajo fue realizado gracias al apoyo proporcionado por CONACyT, mediante los financiamientos núm. 117983 y núm. 28994M
(1) Dirección de Biología Molecular de Patógenos, Centro de Investigación sobre Enfermedades Infecciosas, Instituto Nacional de Salud Pública, México.
* Smad: contracción de los genes que codifican para las proteínas SMA/MAD. MAD: un gen de drosophila (mother against dpp), SMA: proteína homóloga a MAD en eucariores superiores.
** Datos no publicados
Fecha de recibido: 3 de agosto de 2000 · Fecha de aprobado: 16 de enero de 2001
Solicitud de sobretiros: Mtro. Oscar Peralta Zaragoza. Dirección de Biología Molecular de Patógenos, Centro de Investigación sobre Enfermedades Infecciosas, Instituto Nacional de Salud Pública. Avenida Universidad 655, colonia Santa María Ahuacatitlán, 62508 Cuernavaca, Morelos, México. Correo electrónico: operalta@correo.insp.mx


{kind=link}
{kind=link}