











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La trisomía 18 (T18) o síndrome de Edwards (SE) se caracteriza por retraso del crecimiento prenatal, rasgos craneofaciales característicos, esternón corto, postura distintiva de las manos con dedos sobrepuestos, hipoplasia ungueal, hallux corto, así como malformaciones mayores, de las cuales las principales son del corazón.1
La T18 afecta a uno de cada 5,000 recién nacidos (RN) vivos y es la segunda cromosomopatía autosómica más frecuente en el humano, superada sólo por el síndrome de Down.1,2 En alrededor de 95% de los casos el SE se produce por una T18 regular, y son infrecuentes otros cariotipos como los mosaicos, trisomías parciales o translocaciones.3
En general el pronóstico de estos pacientes es malo, ya que la expectativa de vida de quienes no reciben tratamiento en cuidados intensivos es de dos a 10 días.2,4
Estos pacientes pueden presentar malformaciones genitales en 3-8% de los casos y anomalías de las vías urinarias en 14-21%.5,6 La anomalía de prune-belly (APB), conocida también como síndrome de Eagle-Barrett o de la tríada (músculos abdominales flácidos, anomalías del tracto urinario y criptorquidia) es una malformación genitourinaria que se ha reportado excepcionalmente en el SE.7 El objetivo del presente trabajo es informar sobre un RN masculino con SE y APB.
Presentación del caso
Se trata de un RN producto de la tercera gesta de madre de 26 años y padre de 27 años. Su primer embarazo resultó en un aborto de seis semanas, y el segundo fue un varón sano. La madre tiene antecedente de colecistectomía y el padre de hipoplasia renal derecha y reflujo vesicoureteral (RVU). Además, la tía abuela y el tatarabuelo, por rama paterna, fueron referidos también con RVU.
La ultrasonografía al quinto mes de gestación identificó restricción del crecimiento, polihidramnios, megavejiga y valvas uretrales posteriores. Nació por cesárea a las 31 semanas de gestación con peso de 725 g (< P3), talla de 30 cm (< P3), perímetro cefálico de 26 cm (< P3). Apgar de 6-4-2, por lo que requirió administración de presión positiva continua de la vía aérea. Sin embargo, el bebé persistió en malas condiciones respiratorias y hemodinámicas, por lo que falleció cuatro horas después de su nacimiento.
En la exploración física se encontró occipucio prominente, hipertricosis frontociliar, blefarofimosis, puente nasal deprimido, pabellones auriculares simplificados, cuello corto (Figura 1A), piel redundante en nuca, esternón corto, pezones hipoplásicos, abdomen flácido con piel delgada y arrugada (prune-belly), defecto en línea media infraumbilical de aspecto cicatrizal, escroto hipoplásico y criptorquidia bilateral (Figura 1B), además de clinodactilia de quintos dedos, con pliegue único de flexión, pliegues digitales distales borrados, pliegues palmares aberrantes, retroflexión de primer dedo del pie, uñas cortas, calcáneos prominentes y pie equinovaro.
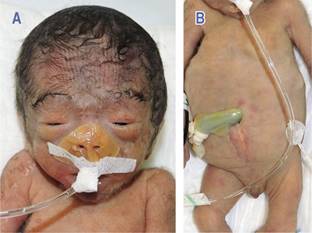
Figura 1: A) Fenotipo facial del paciente típico de síndrome de Edwards, que incluye diámetro bifrontal estrecho, hipertricosis frontociliar, telecanto y fisuras palpebrales estrechas. B) También se observa esternón corto, pezones hipoplásicos, abdomen flácido con piel delgada y arrugada (prune-belly), defecto de la línea media infraumbilical de aspecto cicatrizal, hipoplasia de escroto y criptorquidia bilateral.
La radiografía de tórax mostró 11 pares costales. No se realizaron estudios adicionales de imagenología y los padres no desearon la realización de autopsia.
Se realizó cariotipo en sangre periférica con un nivel de resolución de 450 bandas, el resultado fue: 47,XY,t(5;20)(q22;p11.2),t(8;18)(q22;q11.2),+18 (Figura 2A). Mientras que por microarreglos basados en hibridación genómica comparativa (aCGH), con resolución 60K se encontró: arr(18)×3 (Figura 2B), lo que indicó ganancia de copia simple de todo un cromosoma 18 (Figura 2C); el patrón genómico corresponde a sexo masculino, sin detectar ninguna otra pérdida o ganancia genómica.

Figura 2: A) Cariotipo en sangre periférica con resolución de 450 bandas con resultado: 47,XY,t(5;20)(q22;p11.2),t(8;18)(q22;q11.2),+18, que muestra una doble translocación recíproca aparentemente balanceada, una entre los cromosomas 5 y 20 (cabeza de flecha negra) y otra entre el 8 y 18 (cabeza de flecha blanca), además de un cromosoma 18 adicional completo (recuadro rojo). B) Estudio de microarreglos basados en aCGH que identificó (recuadro rojo) la duplicación patogénica del cromosoma 18. C) Imagen en detalle del cromosoma 18, en donde se observa duplicación patogénica en las citobandas 18p11.32q23, coordenadas genómicas chr18:64,647-78,010,173, que corresponden a una trisomía 18 completa.
Aunque los padres no desearon que se llevaran a cabo más estudios genéticos, se brindó asesoramiento genético.
Los procedimientos diagnósticos realizados se llevaron a cabo en cumplimiento a los principios establecidos en la declaración de Helsinki, en los de la Ley General de Salud de México, así como con las normas del Comité Científico y de Ética de nuestro hospital.
Discusión
En el caso que presentamos, la detección prenatal de megavejiga y valvas uretrales posteriores, junto a los hallazgos de abdomen flácido con piel delgada y arrugada, así como criptorquidia bilateral confirmaron la presencia de APB.
Con respecto a la “atrofia cicatrizal”, observada en la línea media infraumbilical, se debe considerar como hallazgo inusual (Figura 1B), ya que no hay reportados en la literatura de pacientes con APB con una lesión similar. Suponemos que puede ser atribuible a una posible ruptura del abdomen fetal que cicatrizó in utero, la cual ocurrió por la distensión abdominal originada por la megavejiga.
La APB tiene una prevalencia de 3.8 por cada 100,000 recién nacidos vivos.8 Se han propuesto dos principales teorías patogénicas para su ocurrencia, la primera se basa en la obstrucción distal del tracto urinario de inicio temprano (atresia uretral, obstrucción severa por válvulas uretrales posteriores o dilatación prostática fetal) que obstaculiza la salida de orina, originando distensión progresiva de la vejiga (megavejiga) como del abdomen, lo cual altera el desarrollo de músculos abdominales y obstruye el descenso de los testículos.8-10 La segunda teoría es por afección del desarrollo mesenquimal, por un defecto de migración de mesoblasto, entre la semana seis y siete de gestación, que causa deficiencia en la formación de los músculos abdominales, conductos mesonéfricos, paramesonéfricos y órganos urinarios.11 En nuestro caso, la evidencia prenatal de megavejiga y valvas uretrales posteriores parece corresponder a la primera de estas teorías.
Un punto para destacar es que en la mayoría de los embarazos con productos que presentan APB se determina que hay oligohidramnios, como consecuencia de la obstrucción uretral.12 Esto contrasta con nuestro caso, ya que se identificó polihidramnios, el cual atribuimos a una deglución anormal in utero, que es un problema típico en los fetos con T18.1
Se ha descrito que la T18 es la anomalía cromosómica que más frecuentemente se asocia a megavejiga y APB.7,13-19 Al revisar la literatura, solamente encontramos un reporte de un RN vivo de T18 con APB, que falleció a los siete días de vida y en cuya autopsia se identificó riñón en herradura, dilatación ureteropélvica y papilomas múltiples en vejiga.7 Otros reportes de casos similares provienen de series en los que se realizó terminación del embarazo por megavejiga o uropatía obstructiva.13-19
El cariotipo 47,XY,t(5;20)(q22;p11.2),t(8;18)(q22;q11.2),+18 de nuestro paciente, corresponde a una T18 regular con dos translocaciones recíprocas simples independientes, una entre los cromosomas 5 y 20, y la otra entre los cromosomas 8 y 18, ambas aparentemente balanceadas (Figura 2A). Sólo en 0.2% de los casos de T18 se puede encontrar una translocación coincidente que no contribuye al fenotipo,20 por lo que consideramos este hallazgo como único, por ser portador de dos translocaciones recíprocas aparentemente balanceadas. Este cariotipo corresponde a un rearreglo cromosómico complejo (RCC), clasificado como intercambio de dos vías “doble”, debido a que representa la coincidencia de dos translocaciones recíprocas simples separadas, lo cual significa una forma más simple de RCC, al comparar con intercambios de tres vías, que son translocaciones con intercambio de material que involucran a tres cromosomas.21 En el análisis con aCGH confirmó la T18, pero sin identificar desbalances genómicos adicionales (Figura 2B y C). Aunque no pudimos conocer el cariotipo de sus padres, se sabe que la mayoría de estas translocaciones recíprocas “dobles” ocurren de novo.22 Con este mismo análisis, se descartaron deleciones, duplicaciones o amplificaciones adicionales, para un nivel de resolución de aproximadamente 100 kB. Sin embargo, no se logró descartar desbalances genómicos de menor tamaño, u otros defectos genómicos en los sitios de ruptura del RCC, los cuales se detectan a través de estudios de secuenciación de siguiente generación (NGS, por sus siglas en inglés), como la disrupción de genes, el efecto de posición, la disrupción de dominios topológicamente asociados, o la presencia de variantes de nucleótido sencillo relacionadas o no a los sitios de ruptura.23
A fin de precisar el origen de la APB; se analizaron los genes localizados en los sitios de ruptura identificados en las dos translocaciones recíprocas presentes en nuestro caso, sólo llamó la atención el gen GREB1L, localizado en 18q11.2, por ser causante de hipoplasia o aplasia renal, RVU, hidronefrosis o inclusive atresia uretral con secuencia de Potter.24 Otros genes causales de APB como FLNA, HNF1B, MYOCD, SOX17, CHRM3, ACTA2 y ACTG2 no guardan relación topológica con los sitios de ruptura involucrados en las dos translocaciones recíprocas simultáneas.10,12 Sin embargo, los reportes previos apoyan más el que la APB pueda estar totalmente explicada sólo por la T18 regular.7,13-19

