











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las variantes en el número de copia (CNV, por sus siglas en inglés) son alteraciones estructurales como duplicaciones, deleciones, inversiones y traslocaciones que se localizan en diferentes regiones genómicas. Se han identificado diferentes CNV en 14% de los pacientes con trastornos del neurodesarrollo1 y en 3-25% de los pacientes con cardiopatías congénitas.2 Aunque no todos los CNV son causantes de fenotipos patogénicos, los CNV contribuyen a un gran número de síndromes de microdeleción y microduplicación con fenotipos heterogéneos. Con el uso de nuevas tecnologías diagnósticas como los microarreglos cromosómicos, la detección de CNV en distintas regiones genómicas está en aumento.3
Las CNV comúnmente se observan en los rearreglos cromosómicos complejos (RCC). Los RCC son alteraciones que involucran más de dos puntos de ruptura que modifican la estructura cromosómica.4 Algunos autores han postulado que puede existir una alteración en la composición de más de dos cromosomas; sin embargo, otros autores han identificado casos de RCC que modifican de forma extrema la composición de un solo cromosoma.4 Los RCC pueden ser inserciones-traslocaciones, inversiones asociadas a CNV, traslocaciones con más de dos cromosomas, o bien, la combinación de cualquiera de las anteriores.4 Los RCC se pueden originar de novo o ser heredados en casos familiares.4 Sin embargo, no hay un consenso para clasificar a los RCC debido a su complejidad y a nuevos descubrimientos basados en el avance de la tecnología.4,5
El fenotipo de los individuos con RCC es altamente variable, ya que depende del cromosoma afectado, la región, tamaño de la pérdida o ganancia de material, la región genómica implicada y la coexistencia con otros CNV, por lo que cada caso es único.4
El objetivo es presentar un paciente con trastornos del neurodesarrollo, múltiples dismorfias y cardiopatía congénita causadas por un RCC.
Presentación del caso
Paciente femenino de cinco años siete meses referida a la consulta del Servicio de Genética del Centro de Rehabilitación e Inclusión Infantil Teletón (CRIT) en Michoacán por trastornos en el neurodesarrollo y detección de malformaciones.
En cuanto a los antecedentes heredofamiliares (Figura 1): madre de 34 años con diagnóstico de epilepsia bajo tratamiento, y con dos abortos previos. Padre de 34 años, aparentemente sano. Medio hermano materno de 12 años con trastorno por déficit de atención e hiperactividad y epilepsia (pero sin valoración por el servicio de genética). Se negó consanguinidad y endogamia. Como antecedentes perinatales, la madre cursó con preeclampsia, se suspendieron antiepilépticos, y se negó exposición a otros teratógenos. Ecografías fetales sin alteraciones. La paciente nació por parto eutócico a las 37 semanas de gestación.

Figura 1: Genealogía. Propósito III-1: 46,XX,add(18)(p11.2); arr[GRCh37] 7p22.3(43360_705271)x3, 17q25.3 (78763768_81041938)x3, 18p11.32p11.31(136226_4642999)x1. Padre II-1: 46,XY; Madre II-2: 46,XX.
Al nacimiento se detectaron dismorfias faciales y un soplo cardiaco, por lo que fue referida a un hospital pediátrico. En dicho hospital se le realizó resonancia magnética de encéfalo que reveló una atrofia cortical. También se le solicitó un cariotipo, el cual fue reportado sin alteraciones (46, XX). Durante su hospitalización, se diagnosticó defecto septal atrial tipo ostium secundum por cardiología pediátrica. Fue referida a un hospital de tercer nivel, indicando terapia de estimulación temprana.
En servicio de neurología pediátrica del hospital de tercer nivel se dio seguimiento por retraso global del desarrollo, mientras que por cardiología se indicó tratamiento farmacológico cardiovascular. Hasta los dos años ocho meses la paciente continuó con terapia de estimulación temprana, que fue reemplazada por un programa con ejercicios de psicomotricidad.
A los tres años, en tomografía axial computarizada de cráneo se reveló atrofia córtico-subcortical, y se brindó tratamiento quirúrgico para corrección de estrabismo. En el servicio de genética se solicita nuevamente un cariotipo, en el cual se detectó material adicional en el brazo corto del cromosoma 18 (46, XX, add (18) (p11.2)), realizando cariotipo a los padres (46, XX y 46, XY).
Dado que la paciente continuaba con problemas de la marcha, aprendizaje y del lenguaje, los padres acuden al CRIT para iniciar rehabilitación, a la edad de cinco años siete meses. En el servicio de rehabilitación se identificó que la paciente presenta retraso en los hitos del desarrollo psicomotor: sostén cefálico (a los seis meses de edad), pinza gruesa (dos años) y fina (tres años), sedestación (dos años seis meses), gateo (tres años seis meses), marcha (cuatro años) y control de esfínteres (cinco años). Comunicación humana observó retraso alálico del lenguaje, mientras que por neurología pediátrica y paidopsiquiatría se diagnostica trastorno motor y del lenguaje, así como discapacidad intelectual leve.
En la consulta de genética se obtiene la siguiente información: peso 13.7 kg (z -3.1), talla 99 cm (z -2.9), perímetro cefálico de 47 cm (z -2.9), microcefalia, estrechamiento bitemporal, implantación del cabello anterior y posterior baja, frente amplia y prominente, fisuras palpebrales oblicuas hacia arriba, epicanto bilateral, leve ptosis palpebral, cejas anchas y dispersas, puente nasal deprimido, base de la nariz ancha, filtrum largo y poco marcado, hipoplasia malar bilateral, pabellones auriculares de baja implantación, paladar alto, úvula con punta bífida, labio superior delgado, ruidos cardiacos rítmicos, soplo diastólico, escoliosis dorsolumbar, tono motor disminuido, trofismo disminuido, lenguaje monosilábico y voz con timbre hiperrinofónico.
Al revisar el cariotipo tomado a los tres años, junto con los antecedentes familiares y datos clínicos, se solicita microarreglo cromosómico. El estudio se realizó por medio de un microarreglo CytoSCan HD, en el cual se detectaron tres variantes estructurales patogénicas, dos amplificaciones de 662 kilobases (kb) arr[GRCh37] 7p22.3(43360_705271)x3 y 2.2 megabases (Mb) arr[GRCh37] 17q25.3(78763768_81041938)x3 y una deleción de 4.5 Mb arr[GRCh37] 18p11.32p11.31(136226_4642999)x1 (Figura 2). Posterior a la obtención de los resultados se brindó asesoramiento genético a los padres.
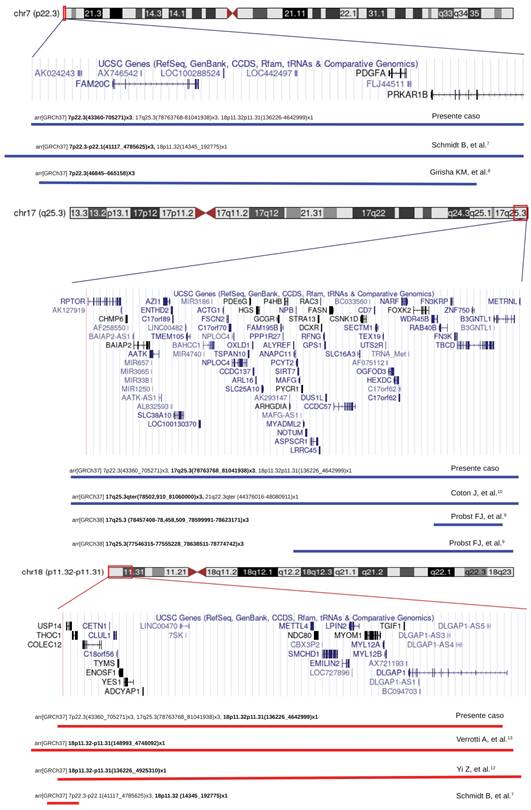
Figura 2: Mapa genético (genes codificantes y no codificantes) en las regiones genómicas de las citobandas 7p22.3, 17q25.3 y 18p11.32-p11.31. Las líneas azules corresponden a las regiones duplicadas y las líneas rojas corresponden a las regiones deletadas, reportadas en casos previos3-5,7,9,10 y en el presente caso. Imagen modificada de la herramienta UCSC Genome Browser Custom Tracks Tool (https://genome.ucsc.edu; Feb 2009 versión Feb. 2009 [GRCh37/hg19]).
Ya con el diagnóstico genético, se decide mantener manejo multidisciplinario con énfasis en las alteraciones neuroconductuales y cardiopatía congénita. El plan de rehabilitación incluyó apoyo pedagógico, terapia física, del lenguaje, ocupacional y pulmonar, además fue integrada a un programa de inclusión. La paciente logró marcha independiente, adquisición de lenguaje y una inclusión escolar. Sin embargo, la paciente fallece a los 10 años por complicaciones tras la corrección quirúrgica de la cardiopatía congénita.
Discusión
El diagnóstico de los pacientes con RCC puede resultar un desafío, principalmente porque la presentación clínica suele ser única e inespecífica, como en el caso que se expone. Pero es de resaltar que el trastorno del neurodesarrollo dio la pauta para buscar etiología cromosómica. A la paciente le fue realizado cariotipo en dos ocasiones, una de ellas sin alteraciones (al nacimiento) y otro mostrando solamente material adicional del brazo corto del cromosoma 18. Si bien hubo un reconocimiento temprano de una posible entidad sindrómica, el cariotipo resultó con limitaciones para un diagnóstico preciso. La gran mayoría de los casos con RCC no se detectan por citogenética convencional, en gran medida porque la resolución del cariotipo es insuficiente para detectar CNV menores de 5 Mb. En cambio, el microarreglo cromosómico fue de gran ayuda para identificar los cambios submicroscópicos no visibles por citogenética convencional.3
El microarreglo cromosómico tiene utilidad clínica; por ejemplo, en la actualidad sirve como primera línea para el diagnóstico de trastornos del neurodesarrollo y de otras anomalías congénitas. Pero se debe tomar en cuenta que en trastornos del neurodesarrollo, el rendimiento diagnóstico es variable alcanzando un máximo de 28%. Por lo que, en ocasiones, debe acompañarse de otros estudios diagnósticos o extender los estudios a los familiares para complementar el diagnóstico.3
La coexistencia de tres CNV patogénicas en la paciente (dos microduplicaciones subteloméricas [7p22.3 y 17q25.3] y una microdeleción subtelomérica [18p11.32-18p11.31]), detectadas por el microarreglo cromosómico, sugiere un RCC por una translocación no balanceada. No obstante, no fue posible realizar una mayor caracterización para determinar si la traslocación involucra los tres CNV detectados en la paciente t(7; 17; 18), o si se trata de una traslocación t(17;18) más una microduplicación pura 7p22.3. Otro punto por analizar es que, para comprender el origen de un RCC y brindar un asesoramiento genético preciso, es necesario extender los estudios genéticos a los familiares. En el presente caso no se logró identificar alteraciones cromosómicas por medio de cariotipo en los padres; sin embargo, en la genealogía se observa una posible trasmisión por rama materna de un fenotipo con alteraciones del neurodesarrollo. El antecedente materno de epilepsia, abortos recurrentes e hijo con fenotipo similar a la paciente sugieren que la madre podría ser portadora de un RCC. Para determinar el origen en estos casos se realizan estudios de hibridación in situ fluorescente (FISH) con sondas subteloméricas o microarreglos cromosómicos en los familiares directos, lo cual no se realizó en la paciente que presentamos.
Los mecanismos en la génesis de un RCC en pacientes con anomalías congénitas son complejos.5 Se ha propuesto la cromoanagénesis, donde un solo evento catastrófico da lugar a rupturas cromosómicas, seguido de un reordenamiento caótico de los fragmentos cromosómicos, alterando la constitución normal de uno o más cromosomas, dando como resultado CNV y otras alteraciones estructurales.5 El término agrupa tres fenómenos distintos: cromotripsis, cromoanasíntesis y cromoplexia. Cada uno con características propias con relación al mecanismo de reparación de rupturas de doble cadena y firmas moleculares únicas.4,5
Actualmente se está investigando si otros estudios podrían ayudar a comprender la formación, complejidad y mecanismos implicados en un RCC de forma más precisa, tales como la secuenciación de genoma completo y algoritmos bioinformáticos diseñados específicamente para detectar RCC, o bien, por medio de estudios de transcriptoma. Es posible que en el futuro estos métodos sustituyan a los microarreglos cromosómicos.6
Con respecto al análisis de los genes afectados con las características clínicas del paciente, es decir, la correlación genotipo-fenotipo en el presente caso, se sabe que las microduplicaciones subteloméricas del brazo corto del cromosoma 7 (7p)7,8 y brazo largo del cromosoma 17 (17q),2,9-11 así como las microdeleciones subteloméricas del brazo corto del cromosoma 18 (18p)7,12-14 son poco frecuentes de forma pura, pero la coexistencia de éstas en un mismo paciente se considera raro. En el caso que presentamos, a pesar de que los CNV se encuentran en regiones genómicas distintas, las características de la paciente (fenotipo) ya se han relacionado con ciertos CNV como la discapacidad intelectual, talla baja, alteraciones en el sistema nervioso central (SNC), malformaciones cardiacas, dismorfias craneofaciales (microcefalia, ptosis, estrabismo, paladar alto, labios delgados, entre otras), y alteraciones musculoesqueléticas (Tabla 1).2,7-14 Los reportes en regiones genómicas similares a las detectadas en la paciente se han presentado como translocaciones no balanceadas y CNV puras (Tabla 1 y Figura 2).2,7-10,12,13 Al haber una coexistencia de CNV con fenotipos similares es difícil discernir cuál de ellos tuvo mayor representación. El fenotipo combinado también se ha observado en otros casos de translocación no balanceada que involucran CNV en regiones genómicas similares (Tabla 1 y Figura 2).7,10
Tabla 1: Fenotipo presente en casos clínicos previamente reportados de CNV en las regiones genómicas 7p22.3, 17q25.3 y 18p11.32-p11.312,7-10,12,13 y presente caso.
| Duplicación pura | Deleción pura | Traslocaciones no balanceadas | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 7p22.3 | 17q25.3 | 17q25.3 | 17q25.3 | 18p11.32-p11.31 | 18p11.32-p11.31 | t(7;18) | t(17;21) | t(17;18) dup(7p22.3) versus t(7;17;18) | |
| Girisha KM et al.(8) | Probst FJ, et al.(9) | Probst FJ, et al.(9) | Monteiro RAC, et al.(2) | Verrotti A, et al.(13) | Yi Z, et al.(12) | Schmidt B, et al.(7) | Coton J, et al.(10) | Presente caso | |
| Trastorno del neurodesarrollo | |||||||||
| Retraso global del desarrollo | - | + | + | - | - | - | - | - | + |
| Discapacidad intelectual | - | - | + | - | - | - | + | - | + |
| Trastorno motor | - | - | - | - | + | - | + | - | + |
| Trastorno del lenguaje | - | - | - | - | + | - | + | + | + |
| Problemas del aprendizaje | - | - | - | - | - | - | - | + | - |
| TDAH | - | - | + | - | - | - | - | + | MH |
| Sistema nervioso central | |||||||||
| Epilepsia | - | - | - | - | + | - | - | - | M/MH |
| Alteraciones del cuerpo calloso | - | + | - | - | - | - | - | - | - |
| Atrofia córtico- subcortical | - | - | - | - | - | - | - | - | + |
| Holoprosencefalia | - | - | - | - | - | + | - | - | - |
| Encefalomalacia | - | - | + | - | - | - | - | - | - |
| Hipotonía | - | - | + | - | - | - | - | - | + |
| Alteraciones craneales | |||||||||
| Microcefalia | - | - | + | - | - | - | - | - | + |
| Sinostosis craneales | - | - | + | - | - | - | - | - | - |
| Cardiopatía congénita | |||||||||
| Defectos septales atriales | - | - | - | + | - | - | - | - | + |
| Tetralogía de Fallot | - | - | - | - | - | + | - | - | - |
| Hipoplasia del ventrículo izquierdo | - | - | - | + | - | - | - | - | - |
| Interrupción del arco aórtico | - | - | - | + | - | - | - | - | - |
| Estenosis de la válvula aórtica | - | - | - | - | - | - | + | - | - |
| Arco aórtico pequeño | - | - | - | - | - | - | + | - | - |
| Coartación aórtica | - | - | - | - | - | - | + | - | - |
| Alteraciones óseas | |||||||||
| Escoliosis | + | - | - | - | - | - | - | - | + |
| Pectus carinatum | + | - | - | - | - | - | - | - | - |
| Alteraciones óseas en extremidades | + | - | - | - | - | - | - | + | - |
| Contracturas | - | + | - | - | - | - | - | - | - |
| Alteraciones oftalmológicas | |||||||||
| Estrabismo | - | - | - | - | - | - | - | + | + |
| Ptosis palpebral | - | - | - | - | - | - | - | - | + |
| Cataratas congénitas | - | + | - | - | - | - | - | - | - |
| Facies | |||||||||
| Baja implantación de cabello posterior | - | - | - | - | - | - | - | + | + |
| Pabellones auriculares de implantación baja | + | - | - | - | - | - | - | - | + |
| Cejas anchas y escasas | - | - | - | - | - | - | - | + | + |
| Fisuras palpebrales hacia arriba | - | + | - | - | - | - | - | - | + |
| Epicanto | - | - | - | - | - | - | - | + | + |
| Hipoplasia malar | - | - | - | - | - | - | - | ||
| Base de la nariz ancha | + | - | - | - | - | - | - | - | + |
| Filtrum largo | - | - | - | - | - | - | - | + | + |
| Paladar alto | - | - | + | - | - | - | - | - | + |
| Labio y paladar hendidos | - | - | - | - | - | + | - | - | Úvula bífida |
| Otras | + | + | + | - | - | + | - | + | + |
| Otros | |||||||||
| Peso y talla bajos | - | - | - | - | - | - | - | - | + |
| Poliesplenia | - | + | - | - | - | - | - | - | - |
| Hipogonadismo | - | - | - | - | - | - | - | - | - |
CNV = variantes en el número de copia; M = madre; MH = medio hermano; TDAH = trastorno por déficit de atención e hiperactividad.
En el caso que nos ocupa, las regiones genómicas en los CNV, 7p22.3, 17q25.3 y 18p11.32-p11.31 comprenden ocho, 83 y 29 genes respectivamente, según UCSC Genome Browser (GRCh37/hg19) (Figura 2).15 Al analizar los casos previos y los genes implicados en los CNV de este caso, se considera que varios genes podrían relacionarse en la presentación del fenotipo (Tabla 2).2,7,9,12-14,16-20 Por ejemplo, los genes TBCD y RAC3 (17q25.3)16 y el gen USP14 (18p11.3)7,13 que se han asociado a trastornos del neurodesarrollo y alteraciones del SNC pueden estar afectando al fenotipo por medio de una ganancia de dosis génica (triplosensibilidad) y una pérdida de dosis génica (haplosuficiencia) respectivamente. En cambio, el gen METRNL (17q25.3),17 que tiene un papel en el desarrollo neuronal, podría contribuir por disrupción de la arquitectura genómica al encontrarse el punto de ruptura en la región promotora del gen. Cabe resaltar que los genes de la región 17q25.3 podrían contribuir a la afectación cardiaca. Pero también en modelos animales se ha observado que los genes EMILIN2,18TGIF112,14,16 y MYOM119 podrían propiciar un desarrollo anormal del corazón en la región 18p11.32-p11.31;7,12 sin embargo, no hay reportes de defectos septales atriales como en este caso. En cambio, como en el presente caso, algunos autores han sugerido que la triplosensibilidad del gen ARHGDIA (17q25.3) podría contribuir en defectos septales atriales, lo cual se ha observado en modelos animales y en pacientes con duplicaciones puras 17q25.3.2,9
Tabla 2: Correlación genotipo-fenotipo. Genes propuestos que contribuyen a los fenotipos: trastorno del desarrollo y alteraciones del SNC,7,13,14,16,17 cardiopatía congénita2,9,12,14,16,(18,19 y estrabismo.20
| Trastorno del neurodesarrollo y alteraciones del SNC | Cardiopatía congénita | Estrabismo | |
|---|---|---|---|
| 7p22.3 | FAM20C (MIM: 611061) | – | – |
| 17q25.3 | TBCD (MIM: 604649) | ARHGDIA (MIM: 601925) | |
| WDR45B (MIM: 609226) | CSNK1D (MIM: 600864) | ||
| RAC3 (MIM: 602050) | ACTG1 (MIM: 102560) | ||
| PYCR1 (MIM: 179035) | P4HB (MIM: 176790) | NPLOC4 (MIM: 606590) | |
| ACTG1 (MIM: 102560) | NPLOC4 (MIM: 606590) | TSPAN10 | |
| NDUFAF8 (MIM: 618461) | MRPL12 (MIM: 602375) | PDE6G (MIM: 180073) | |
| METRNL (MIM: 616241) | DCXR (MIM: 608347) | ||
| SLC16A3 (MIM: 603877) | |||
| UTS2R (MIM: 600896) | |||
| 18p11.32-p31 | TGIF1 (MIM: 602630) | EMILIN2 (MIM: 608928) | – |
| USP14 (MIM: 607274) | TGIF1 (MIM: 602630) | ||
| MYOM1 (MIM: 603508) | |||
| TYMS (MIM: 188350) | |||
| MYL12A, MYL12B (MIM: 609211) | |||
| LPIN2 (MIM: 605519) |
SNC = sistema nervioso central; MIM = número de identificación del gen en OMIM.16
Al tratarse de una paciente pediátrica, el asesoramiento genético fue dirigido a los padres, el cual se dirige a 1) comprender el diagnóstico y mecanismo patogénico, 2) conocer el pronóstico, 3) obtener información para la toma de decisiones médicas o de la vida, 4) conocer los riesgos de recurrencia, 5) extender pruebas genéticas a otros familiares con riesgo o sospecha, 6) conocer alternativas reproductivas (como el diagnóstico preimplantacional).3 Cabe mencionar que los portadores de RCC tienen un riesgo elevado de tener descendencia con anormalidades cromosómicas similares o a la formación de nuevos RCC.4
Por último, se debe destacar que esta paciente nos hace reflexionar sobre la necesidad del manejo multidisciplinario de pacientes similares, lo cual contribuye a mejorar su calidad de vida y a la de sus familias.
Conclusiones
Los pacientes con RCC pueden presentar múltiples manifestaciones como trastornos del neurodesarrollo, dismorfias y malformaciones cardiacas; su presencia debería alertar sobre su reconocimiento temprano a fin de ofrecer manejo integral incluyendo expertos en genética, quienes elegirán la realización de estudios citogenómicos para llegar al diagnóstico definitivo, lo cual debería ayudar a disminuir la odisea diagnóstica que, con mucha frecuencia, estas familias padecen.

