











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Se reconoce a la holoprosencefalia (HPE) como la anomalía morfológica más común durante el desarrollo del cerebro anterior (también conocido como prosencéfalo), siendo su prevalencia en 1/10,000 nacimientos. La falla completa o incompleta de la división estructural del cerebro en la tercera o cuarta semana de gestación, da lugar a la formación incompleta de ambos hemisferios y de estructuras cerebrales profundas, tractos olfatorios y ópticos.1-3
El espectro clínico es amplio, teniendo desde la muerte fetal hasta pacientes que llegan a la edad adulta, pero con deterioro cognitivo importante. El espectro clínico corresponde a los diferentes subtipos: alobar, semilobar, la variante interhemisférica media (MIH o sintelencefalia) y la septopreóptica.4 La HPE se asocia con anomalías faciales o craneales (agenesia premaxilar, ciclopía, cebocefalia) y sistémicas (cardiopatías congénitas, polidactilia, defectos urogenitales y vertebrales).5,6
Su etiología es considerada heterogénea, ya que se ha relacionado con problemas genéticos, puesto que va desde casos en los que la HPE forma parte de un síndrome, o bien, defectos aislados,4 pero también se considera que puede ser multifactorial, particularmente asociada a diabetes gestacional.1,7-9
La sintelencefalia es una de las variantes menos frecuentes de la HPE, en la cual se involucra la convexidad frontal alta y una fisura interhemisférica deficiente (motivo por el cual recibe su otro nombre, variante interhemisférica media). Por la falta de división de la porción frontal posterior y parietal de los hemisferios, su resultado es hipoplasia o aplasia del tubo neural.10-12
El objetivo de presentar el caso de una recién nacida con sintelencefalia tiene la finalidad de favorecer el reconocimiento de la variante menos común de la holoprosencefalia.
Presentación del caso
Paciente de sexo femenino nacida a las 38.1 semanas de gestación. Su madre de 32 años, nacida en Oaxaca, México, con bachillerato concluido, actualmente residente en Monterrey, Nuevo León, México, de religión protestante. Se negaron antecedentes de alcoholismo, tabaquismo, uso de drogas o tatuajes. Sin embargo, tenía ocho años con diagnóstico de diabetes mellitus tipo 2 tratada con metformina e insulina rápida, e insulina NPH, además de hipertensión arterial sistémica, tratada con alfametildopa. El padre de 36 años, nacido en Oaxaca, con preparatoria completa, trabajador de empresa metalúrgica, sin antecedentes personales de importancia, de religión protestante, niega alcoholismo, tabaquismo, uso de drogas o tatuajes. Ambos tenían un hijo de cuatro años, aparentemente sano. No se identificó consanguineidad o endogamia, o antecedentes familiares de labio/paladar hendido o de cardiopatía congénita.
La paciente fue producto de segunda gestación, con ingesta de hierro y ácido fólico desde la primera semana de embarazo y con adecuado seguimiento prenatal (18 consultas y realización de cuatro ultrasonidos). En la semana 21 se realiza el diagnóstico de HPE por ecografía en un centro privado, motivo por el cual se realiza referencia a nuestro centro.
Se realizó cesárea a las 38 semanas de gestación. Al nacer presentó llanto y respiración espontánea, frecuencia cardiaca mayor a 100, escala Apgar 8/9, peso 2,900 gramos, 45 cm de longitud, 32 cm de circunferencia cefálica. A la EF presentaba labio y paladar hendidos, y pie equino varo, como únicas alteraciones.
Debido al diagnóstico de HPE, inmediatamente posterior a su nacimiento se tomaron imágenes de resonancia magnética, que revelaron anomalías de la línea media caracterizadas por fusión interhemisférica parcial de los lóbulos frontales, asociada con la ausencia de la rodilla del cuerpo calloso, que tiene una longitud de 24.4 mm, cuerpo de 2.3 mm y esplenio de 3.1 mm, condicionando disminución de la amplitud de los huecos frontales, pero con núcleos grises conservados (Figura 1).
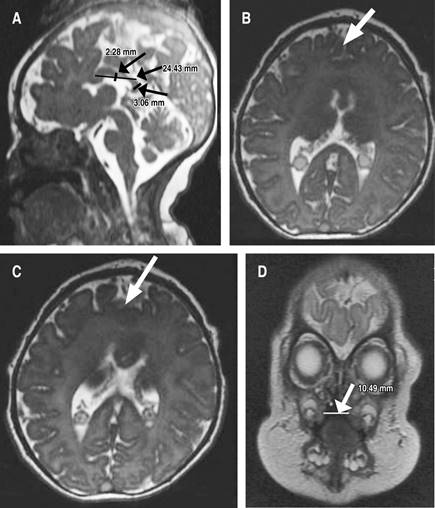
Figura 1: Sintelencefalia demostrada por resonancia magnética nuclear. A) Ausencia de la rodilla del cuerpo calloso, este cuenta con un tamaño de 24.4 mm (normal 5.1 ± 1.0 mm), cuerpo de 2.3 mm (2.3 ± 0.6 mm) y esplenio de 3.1 mm (normal 3.7 ± 0.6 mm), lo que condiciona una disminución en la amplitud del receso frontal, cerebelo y tallo encefálico de morfología e intensidad preservada. Sistema infratentorial y espacio subaracnoideo normal. B y C) Anormalidades de línea media caracterizadas por fusión parcial de los lóbulos frontales. D) Ausencia en la fusión del paladar duro que comunica con la cavidad nasal derecha, ausencia de fusión de labio superior derecho, defecto de 10.5 mm. Nota: estudio realizado el día del nacimiento de la paciente.
A los dos días de nacimiento, un ultrasonido renal demostró ambos riñones disminuidos de tamaño, aumento de ecogenicidad en corteza y ectasia pielocalicial bilateral. Por desaturación de oxígeno persistente se efectuó ecocardiograma, encontrando corazón univentricular, con válvula auriculoventricular única y atrésica, comunicación interventricular, trasposición de grandes vasos, estenosis valvular pulmonar grave y persistencia de conducto arterioso.
Ingresó en la Unidad de Cuidados Intensivos por continuar con saturación de oxígeno entre 58-75%, sin datos de dificultad respiratoria, pero con cianosis generalizada y soplo audible grado 2. Luego de dos semanas en cuidados intensivos presentó bradicardia y posteriormente paro cardiorrespiratorio. Fallece por choque cardiogénico.
Es conveniente señalar que el cribado o tamiz metabólico neonatal reportó elevación de 17-hidroxiprogesterona, y que no se llevó a cabo el abordaje desde el punto de vista genético.
Discusión
En 1987, DeMyer clasificó a HPE en tres variantes, de acuerdo con su gravedad: alobar, semilobar y lobar. A las cuales posteriormente se agregó la variante interhemisférica media, así como la septopreóptica o displasia septopreóptica.12,13
La variante alobar es considerada la de peor pronóstico, con una completa o casi completa falta de separación de los hemisferios cerebrales, un único ventrículo en el prosencéfalo y, usualmente, con comunicación con la cisterna dorsal. Los pacientes con ésta pueden presentar hipotonía y convulsiones al nacer. Por su parte, en la variante semilobar, los hemisferios anteriores no se separan y son pequeños; además los cuernos frontales de los ventrículos laterales y la porción anterior del cuerpo calloso, por lo general, están ausentes. En estos pacientes las malformaciones faciales son pocas o no existen, pero tiene retraso en el desarrollo, principalmente en las habilidades motoras. Mientras que la variante lobar es la menos grave; únicamente las porciones más rostrales/ventrales de la neocorteza no están separadas y, el rostrum y el genu del cuerpo calloso pueden estar ausentes.
La sintelencefalia es una variante menos grave que las descritas previamente, y con anatomía diferente. La regiones menos separadas se localizan basalmente en el cerebro anterior. Lo que falla en separarse son las porciones frontales y parietales del lóbulo frontal; además, el cuerpo calloso usualmente está ausente, mientras que la rodilla y esplenio están formados. Por último, en la variante septopreóptica hay una ausencia completa del septum pellucidum, hipoplasia de los nervios y quiasma óptico.14-16
La HPE ocurre durante la cuarta o quinta semana de gestación. En este periodo, la más anterior de las tres vesículas cerebrales primarias tiene una división horizontal formando el telencéfalo (que dará lugar a los hemisferios cerebrales y a los ventrículos laterales) y en diencéfalo (que formará tálamos, neurohipófisis y tercer ventrículo). A esta división se denomina clivaje y, cuando falla, origina cualquiera de las diferentes HPE.17
Como se mencionó, la etiología de HPE puede ser genética o multifactorial. De esta última, la más frecuente es por embriopatía diabética, la cual se asocia a malformaciones del sistema nervioso central, incluyendo a la anencefalia, encefalocele y HPE, así como a malformaciones craneofaciales (microsomia hemifacial, microtia, labio y paladar hendidos). Además, se ha relacionado a malformaciones cardiovasculares y esqueléticas.18-24 Lo anterior resulta relevante para el caso de la paciente que presentamos por el antecedente materno de diabetes mellitus de ocho años de duración, y porque además de la HPE, se identificó una cardiopatía cardiaca compleja, defectos genitourinarios y anomalías orofaciales.
Con respecto a la posible etiología genética, en el año 2010, Kagan y colaboradores25 reportaron que 65.9% de fetos con HPE contaban con cariotipo alterado, de los cuales 86% correspondieron a trisomía 13 o síndrome de Patau. En Argentina, Petracchi y su equipo,26 en 2011, informan una prevalencia de hasta 43% de trisomía 13, como la causa más común HPE; señalando que los pacientes también presentaban microftalmia, anoftalmia, labio y paladar hendidos, polidactilia y pie en balancín. También la HPE se ha asociado con las trisomías 18, 21 y 22.27
También se ha estudiado que hasta en 25% de las HPE puede identificarse una mutación genética, y de los genes implicados son principalmente a ZIC2 (gen involucrado en el desarrollo del cerebro anterior), SHH, SIX3, FGFR1 y FGF8. Otros genes que aún no tienen un rol definido son PTCH1 y TGIF1.27,28 Por último, debe tenerse en cuenta que las HPE puede ser parte de los siguientes síndromes: deleción 13q, 18p, SLOS, Hartsfield, Steinfeld, Culler-Jones.27,29
Conclusiones
La sintelencefalia, cuando aparece de forma aislada, puede hacer que los pacientes tengan pronóstico favorable, pero se ensombrece cuando se acompaña de otras malformaciones mayores. Por lo anterior, el manejo deberá ser multidisciplinario, poniendo especial atención en la participación de especialistas de Genética.

