











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las mutaciones del gen HNF1β fueron descritas por primera vez en 1997 como una causa genética rara de diabetes tipo MODY con presentación en la edad pediátrica, asociándose con un fenotipo multisistémico variable.1,2HNF1β es un factor de transcripción que se requiere para la expresión de numerosos genes, principalmente en el riñón, hígado, páncreas y paratiroides.2-4 De ahí que el fenotipo relacionado con las mutaciones sea: elevación de enzimas hepáticas, diabetes mellitus, hiperparatiroidismo, malformaciones del tracto urinario y genital, quistes renales, así como enfermedad renal tubulointersticial, manifestándose con hiperuricemia o hipomagnesemia.5,6
Se presentan dos casos de niños portadores de la mutación en el gen HNF1β, quienes tuvieron distinto comportamiento clínico.
Reporte de casos
Caso 1. Paciente femenino de 11 años y ocho meses, con peso de 49.8 kg (percentil 70), talla 143.7 cm (percentil 16), IMC 24.12 (percentil 91). Cuenta con diagnóstico prenatal de riñón multiquístico derecho. Desde su nacimiento inicia control por la consulta de nefrología pediátrica detectándose hipocitraturia. Durante el seguimiento al año de edad se agrega hipocalciuria; a los cuatro años, hipomagnesemia (1.2 mg/dL) y elevación de triglicéridos (512 mg/dL), colesterol (293 mg/dL) y enzimas hepáticas (AST 65 U/L, ALT 94 U/L). A los ocho años, mediante un ultrasonido abdominal se observa un hígado de tamaño y morfología normal, homogéneo y sin lesiones focales. Por las múltiples manifestaciones se solicita estudio genético; el análisis por Multiplex Ligation-dependent Probe Amplification (MLPA) mostró una deleción en heterocigosis en el exón 4 del gen HNF1β; al estudiar a los padres no presentaron afectación, por lo que se considera una mutación de novo. Por la asociación de esta mutación con alteraciones a nivel genital se realiza un ultrasonido pélvico, observándose un ovario izquierdo de morfología conservada y útero normal, aunque no se identifica el ovario derecho, la exploración de genitales es normal. En la actualidad, se encuentra asintomática. Pero por parte de Nefrología recibe aporte de magnesio y citrato de potasio; la función en estadio I KDIGO, cistatina C elevada 1.28 mg/L. El riñón derecho ya no se observa en ultrasonido renal, y el riñón izquierdo mide 9 cm con adecuado grosor cortical.
Dado el riesgo de diabetes tipo MODY, también tiene vigilancia por Endocrinología. En exámenes de laboratorio se ha detectado: insulina 25 µUI/mol y glucemia en ayunas 99 mg/dL; pero la glucemia fue de 235 mg/dL en prueba de tolerancia a la glucosa a las dos horas. Se realizó evaluación durante siete días con sensor transcutáneo de glucemia 24 horas siendo normal, por lo que, sólo requirió asesoramiento de nutrición por dislipidemia.
Caso 2. Masculino de 17 años, con peso de 67.6 kg (percentil 26), talla 169 cm (percentil 10), IMC 23.46 (percentil 48). Desde su nacimiento se detectó ectasia pielocalicial izquierda e hiperecogenicidad de la corteza renal. En los primeros estudios de laboratorio se reporta hipercalciuria, hipocitraturia y defecto en la prueba de concentración renal (683 mOsm/kg). A los tres años, por ecografía renal se observó un quiste cortical derecho (Figura 1). A los cuatro años de edad se detectó hipomagnesemia (1.24 mg/dL). A los 13 años, se diagnosticó con diabetes: por glucosa en ayuno de 162 mg/dL, se realizó prueba de carga de glucosa encontrándose anormal (glucemia pasó de 127 a 282 mg/dL a las dos horas). Nivel de insulina 12.38 µUI/mol. Además, presenta dislipidemia con triglicéridos 283 mg/dL, colesterol total de 237 mg/dL, por lo que inicia manejo con metformina y atorvastatina. Por otro lado, se detectó elevación de alanina-aminotransferasa (175 U/L) teniendo ultrasonido hepático que reveló datos de esteatosis. El paciente no tiene alteraciones a la exploración física de genitales.
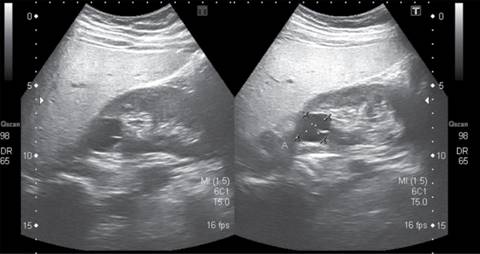
Por esta evolución se sospechó una mutación del gen HNF1β y se solicitó análisis por MLPA. Se demostró una deleción en heterocigosis en los exones 1 a 9 del gen HNF1β, por lo que su diabetes se clasifica como MODY 5. El padre del paciente presenta la misma mutación, pero no tiene manifestaciones clínicas.
Discusión
La mutación del factor nuclear del hepatocito (HNF1β) se ha asociado a malformaciones del riñón y del tracto urinario, afectación tubulointersticial, así como a diabetes y elevación moderada de enzimas hepáticas, hiperuricemia e hipomagnesemia. En la población pediátrica, las alteraciones renales son el fenotipo que con más frecuencia se identifica en los pacientes, mientras que en adultos es por diabetes.7
La afectación del gen puede estar dada por una mutación de novo o esporádica en 50 a 60% de los casos o de forma hereditaria familiar, siguiendo un modelo autosómico dominante.
Las funciones pleiotrópicas de HNF1β como factor de transcripción en varios tejidos (incluido el riñón, páncreas, hígado y el tracto urogenital) dan como resultado las diversas manifestaciones.6 Las manifestaciones como la hipomagnesemia y la pérdida salina obedecen a que el HNF1β interviene en la expresión de varios genes como FXYD2, que codifica la subunidad gamma de la Na+-K+ ATPasa. En condiciones fisiológicas, esta subunidad genera un gradiente de Na+ que permite al cotransportador sensible a tiazidas NCC, el transporte de Na+ desde la luz apical al citosol. Cuando existen mutaciones en el gen HNF1β, se reduce la densidad de proteínas FXYD2 y, por ende, la actividad de la Na+-K+ ATPasa basolateral, lo que conduce a un aumento de la concentración intracelular de sodio que inhibe la actividad del transportador NCC con la consiguiente pérdida salina. El descenso de actividad de NCC afecta el potencial de membrana necesario para la reabsorción apical de magnesio por parte del canal epitelial de magnesio TRPM6. Esta alteración genera hipomagnesemia (Figura 2).2,5,8,9 El defecto de reabsorción de Na+ y Cl- en el túbulo contorneado distal estimula la reabsorción de Na+ en las células principales del túbulo distal y del túbulo colector, se intercambia por K+ con lo que se produce un aumento en la excreción renal de K+. La pérdida de Na+ se compensa, pero no la de Cl-.7 Además, se ha sugerido que el HNF1β regula la transcripción del gen UMOD, por lo que su alteración genera un transporte anormal de uratos con la consiguiente hiperuricemia y gota.

Figura 2: En el túbulo contorneado distal, el Mg2+ se reabsorbe principalmente por rutas transcelulares. Los iones Mg2+ pasan a la célula a través del canal apical TRPM6 y salen de la célula mediante CNNM2 y el intercambiador de magnesio/sodio SLC41A1 (no representado en la Figura). La fuerza apical necesaria para el transporte de Mg2+ es creada por la acción cooperativa de la Na+,K+-ATPasa basolateral, el canal de cloro ClC-Kb, el transportador de K+ Kir 4.1 (involucrado en el síndrome EAST), el cotransportador apical de ClNa sensible a tiazidas NCC, y los extrusores de K+, ROMK (no representado en la Figura) y Kv1.1. Los transportadores afectados en la nefropatía HNF1β se representan con un aspa roja.
Las alteraciones a nivel pancreático se pueden explicar debido a la estructura similar del HNF1β con HNF1α (el gen más afectado en la diabetes MODY tipo 3).
Existe el gen LHX1 en el cromosoma 17q12 contiguo al gen HNF1β, cuya mutación genera otra patología implicada, llamada síndrome de Mayer-Rokitansky-Küster-Hauser.10,11
A diferencia de los dos casos que presentamos, Alvelos y colaboradores describen pacientes con riñones fetales hiperecogénicos y enfermedad glomeruloquística hipoplásica en pacientes neonatales.12 Se ha hecho estudio genético en pacientes con quistes renales detectados de manera prenatal, encontrando 192 variantes de HNF1β intragénico.13
En una investigación realizada en población japonesa que incluía 33 pacientes se observó que la mayoría de los casos tenían anomalías morfológicas en el sistema del tracto urinario-renal, lo cual hace más probable detectar estos casos a través de esta manifestación. La diabetes se desarrolló en 12 casos (38.7%), hiperuricemia e hipomagnesemia se asociaron con seis (19.3%) y 13 casos (41.9%), respectivamente. Hubo malformaciones pancreáticas en siete (22.6%), y 10 pacientes (32.3%) tuvieron alteraciones hepáticas. La tasa de filtración glomerular fue significativamente más baja en los pacientes con variantes heterocigotas en comparación con las de los pacientes que tenían deleción del gen.14

