











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Fraser (SF) es una enfermedad genética de transmisión hereditaria autosómico recesiva cuyo diagnóstico se realiza con base en criterios clínicos. Los criterios mayores incluyen criptoftalmos, sindactilia, anomalías genitales e historia de hermano(a) con SF; mientras que los criterios menores pueden ser malformaciones en oídos, nariz, cavidad oral, laringe, defectos esqueléticos, hernia umbilical, agenesia renal. Se hace diagnóstico con dos criterios mayores y uno menor, o bien, con uno mayor y cuatro menores.1-3
Fue descrito por primera vez en 1962 por George Fraser y se han realizado múltiples revisiones desde entonces.4,5 El SF es una entidad rara; su frecuencia estimada es de 0.2 en 100,000 nacimientos según la serie de 1990-2008 de la Vigilancia Europea de Anomalías Congénitas (EUROCAT).6 Se ha observado que la frecuencia es mayor en padres consanguíneos (15-27%).5,6
El SF es una condición genéticamente heterogénea y, en años recientes, se ha demostrado que existe una gran variabilidad en su presentación. Estudios de familias con SF han reportado mutaciones en el gen FRAS1 ubicado en el cromosoma 4q21, el cual codifica para una proteína de la matriz extracelular.7 Por otro lado, en dos familias en las cuales no existían alteraciones en el FRAS1, se encontró una mutación del gen FREM2 ubicado en el cromosoma 13q13. Se ha descrito que FRAS1 y FREM2 codifican para tres proteínas de la matriz extracelular y para una proteína intracelular de adaptación (GRIP-1). Se ha constatado que tanto FRAS1 como FREM2 tienen la habilidad no sólo de ensamblar los componentes estructurales de la matriz extracelular, sino de modular la actividad de los factores de crecimiento del medio.8 Estas proteínas se requieren para la adhesión epidérmica normal durante el desarrollo in utero; teniendo un papel crucial en el desarrollo embrionario. Estudios en ratones mostraron que la anulación de la proteína GRIP-1 ocasiona varias deformidades en el embrión como agenesia renal, sindactilia y criptoftalmos. El mecanismo a través del cual se producen las malformaciones se debe a la interacción entre GRIP1, FRAS1 y FREM2, puesto que la anulación o la mutación de GRIP1 da como resultado una expresión deficiente de los genes FRAS1 y FREM2.9-11
Se describe un caso de SF que nació en Honduras, dado que hasta ahora no hay casos reportados en Centroamérica.
Presentación del caso
Paciente que nace a las 41 semanas en el Hospital de San Lorenzo, ubicado en la población del mismo nombre, que se ubica en el departamento Valle, en Honduras. El parto fue vaginal, sexo aparente femenino, meconio ++, con circular del cordón al cuello, APGAR 6 y 7, respectivamente. Al nacimiento se detecta dificultad respiratoria, cianosis y múltiples malformaciones, por lo cual se remite al Hospital Escuela Universitario (HEU) de la Universidad Nacional Autónoma de Honduras, ubicado en la ciudad de Tegucigalpa, al sexto día de vida.
El padre de 20 años dedicado a la venta de hamacas y la madre de 18 años es ama de casa, ambos con primaria completa. Negaron consanguinidad; sin embargo, provienen de una aldea pequeña en el Departamento del Paraíso. Historia obstétrica materna: el producto fue de una segunda gestación; siendo el hijo previo sano. Tuvo adecuado control prenatal con enfermera auxiliar en un centro de salud aledaño a su comunidad; afirma infección urinaria en el segundo mes de gestación tratada con amoxicilina, así como leucorrea en las últimas dos semanas de gestación sin tratamiento. Durante el embarazo no se realizó ultrasonido fetal. El tipo sanguíneo de la madre fue O Rh +, serología para VIH negativa.
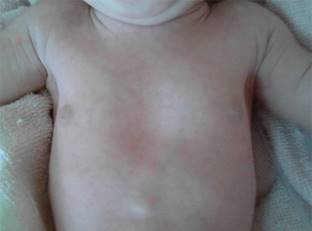
Al momento del ingreso en el HEU la paciente se presenta con frecuencia cardiaca de 148/min, frecuencia respiratoria de 66/min, saturación ambiente de oxígeno en 68%, con estridor inspiratorio. Peso 3.1 kg, talla 51 cm, perímetro cefálico 32 cm. En el examen físico segmentario presenta cabeza con vello en región biparietal, microtia con implantación baja de pabellones auriculares, criptoftalmos derecho completo, mientras que en el lado izquierdo no se identifica globo ocular; coloboma del párpado izquierdo, nariz aplanada, micrognatia (Figura 1), tórax tipo excavatum, teletelia (Figura 2). Asimismo, presenta abdomen con hernia umbilical, en genitales atresia vaginal, hipertrofia del clítoris, ano perforado (Figura 3). En extremidades: sindactilia parcial de manos y pies (Figuras 4 y 5), piel color rosa. Los reflejos del recién nacido están presentes excepto el de prensión.
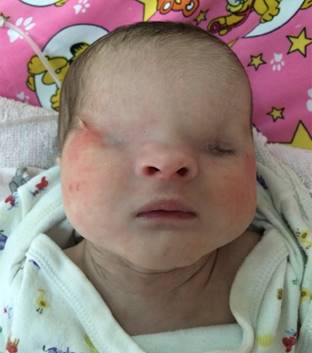
Figura 1: Se observan múltiples malformaciones faciales incluyendo: criptoftalmos, coloboma de párpado izquierdo, remanente en región ocular derecha, nariz aplanada, vello en la región frontoparietal.



Estudios de laboratorio. Hemograma: hemoglobina 15.7 g/dL; hematocrito 44.6%; CHCM 35 g/dL, VCM 105fL, WBC 11,830/mm3; plaquetas 31,100/mm3. Creatina 0.6 mg/dL, BUN 23 mg/dL, bilirrubina total 1.2 mg/dL; bilirrubina indirecta: 0.9 mg/dL. Examen de orina: bilirrubina+; proteínas+; leucocitos 10-12x campo, presencia de hifas con levaduras. Cariotipo: normal 46 XX.
Estudios de imagen. Radiografía de tórax normal. En ecocardiograma: comunicación interventricular de la muscular media de 2 mm y foramen oval permeable. TAC cerebral: ausencia de globos oculares, resto normal. Ultrasonido abdominal: presencia de derivados mullerianos, útero normal, tercio posterior de la vagina presente, riñones sin alteración (Figura 6). Nasofibroscopía: estenosis subglótica.

El manejo durante su estancia hospitalaria fue conservador. A los 21 días de vida ocurre la caída espontánea de muñón umbilical y se empieza a alimentar por vía oral con lactancia. Se trató de corregir la estenosis subglótica; sin embargo, la paciente presentó paro cardiorrespiratorio al mes y dos días de vida y falleció. En los últimos cinco días de vida la paciente se alimentó bien, presentaba signos vitales estables y saturaba al 98%.
Discusión
El SF tiene una amplia variedad clínica, el caso presentado va desde las manifestaciones más frecuentes hasta las menos frecuentes, presentando tres (de un total de cuatro) criterios mayores y cinco criterios menores (de un total de ocho). Según una revisión de 117 casos realizada en 2002, el criptoftalmos se encuentra en 88% de los casos y la sindactilia en 61.5% siendo estos los hallazgos clínicos más frecuentes. La implantación baja de orejas se observó en 53.8% de los casos y la hipertrofia de clítoris fue la anomalía genital más frecuente con 36.8%. La ausencia de cejas y pestañas se aprecia en 29%, el coloboma de párpado en 17.9%, microtia en 16%, atresia del introito vaginal en 12%, aplanamiento nasal en 11%, estenosis subglótica y depresión frontal en 8.5%, anoftalmia en 7% y la hernia umbilical en 6%. Sólo un paciente ha presentado comunicación interventricular.12,13 Las malformaciones cardiacas en SF se mencionan muy poco y se desconoce su frecuencia.14
Con respecto a la consanguinidad, aunque es negada, no se puede descartar por completo debido a que los progenitores vienen de una aldea pequeña donde por lo general los habitantes son parientes en algún grado de consanguinidad.15 En cuanto a los hallazgos del laboratorio al ingreso se atribuyen a una sepsis neonatal. El pronóstico por lo general es malo; sin embargo, es muy variado de acuerdo con el grado en que se manifieste clínicamente el síndrome. Las malformaciones más letales corresponden a la agenesia renal bilateral y a la estenosis laríngea. Aproximadamente el 20% fallece en la primera semana, 13% sobrevive hasta un año y otro 13% sobrevive hasta los 10 años, pero con mala calidad de vida. Durante el embarazo se ha descrito que alrededor del 12% termina en interrupción voluntaria y 6% son prematuros o abortos espontáneos.

