











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink1. Introduction
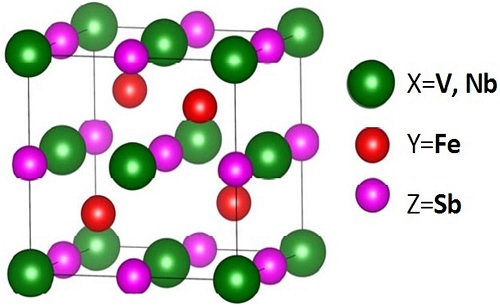
Half-Heusler compounds generally have the composition XYZ [1] and they are ternary alloys with cubic structure where each letter stands for an alloying element, while the complete Heusler are composed according to the pattern X2YZ. By removing one X atom over two, leaving a vacant site in this context, X and Y are transition elements, while Z is an element of the III-V main group. With intermetallic Half-Heusler phase composed of three elements (X, Y, Z) we can obtain semiconductors materials with small-gap and stable structure when we have 18 electrons in the valence band. The narrow band gap at the Fermi level in some of these compounds and voids in the crystal structure are believed to be the key ingredients making these materials promising candidates for thermoelectric applications, they can be used for the manufacture of several optic sensors and solar cells. The Half-Heusler compounds have attracted particular interest first because of their magnetics and their thermoelectric properties at high temperatures [2-3-4]. In this paper, we study ternary Half-Heusler (SH) compounds Fig. 1 which are composed of transition metals with metalloids VFeSb and NbFeSb. The optical and thermal properties are calculated by the full potential linearized augmented plane wave (FP-LAPW) method with the modified Becke-Johnson (mBJ) potential. It is also revealed that the effectiveness of mBJ is based on the proper treatment of the d-orbitals in the highly correlated electron system.
2. Calculation method
In our calculations, we use the Wien2k [5] which is a direct application of the FP-LAPW developed by Blaha, Schwarz and their colleagues [6],and which will allow us to study the structural, electronic and optical properties of Half-Heusler compounds XFeSb: (X=V,Nb).
The algorithm is based on the density functional theory (DFT) within the local density approximation (LDA) proposed by Perdew and Wang [7] for the exchange correlations functional. It calculates the self-consistent solution of the equations of Kohn and Sham [8] who decry the valence electron in a potential created by a periodic array. The electronic configurations for V, Nb, Fe and Sb are V: Z: 23[Ar] 3d3 4s2, Nb: Z: 41 [Kr] 4d4 5s1, Fe: Z: 26 [Ar] 3d64s2 and Sb:Z:51[Kr] 5s24d105p3; In the calculation, the sub shells of the noble gas cores have been distinguished from the sub shells of valence electrons given explicitly.
The k integration over the Brillouin zone is performed using the Monkhorst and Pack mesh [9]. To calculate the band structure of these half-Heusler materials, we used
where
is the electron density,
is the kinetic energy-density,
is the Becke-Roussel (BR) potential, and
was chosen to depend linearly on the square root of the average of |∇ρ|/ρ:
The modified BJ exchange potential yields band gaps with accuracy comparable to other approaches, often orders of magnitude more computationally expensive. The potential we propose is given by Eqs. (1)-(5), where α and β are two free parameters and V cell is the unit cell volume. Minimization of the mean absolute relative error for the band gap of the solids listed in Table II leads to α = −0.012 (dimensionless) and β = 1.023 Bohr 1/2 .
TABLE I Calculated lattice parameter α, bulk modulus B0, and its pressure derivative B’ for VFeSb and NbFeSb.
3. Results and discussion
3.1. Structural properties
Our half-Heusler are cubic ternary alloys with formula XYZ, where X, Y are transition metals, and Z is a sp-valent element. For the calculation of lattice parameter, we will combine Y and Z and X is changed [11]. VFeSb and NbFeSb, like most of half-Heusler ternary alloys, follow a XYZ structure where atom X is located at (0.5, 0.5, 0.5), atom Y at (0.25, 0.25, 0.25), and atom Z at (0, 0, 0) as can be seen in Fig. 1. To start our calculations we have used the experimental lattice constant. In Table Iwe summarize our calculated lattice parameter α and bulk modulus B 0 along with its pressure derivative B’ for VFeSb and NbFeSb with the LDA. The lattice constants and bulk modulus are computed by fitting the total energy versus volume according to the Murnaghan’s equation of state [12].
When we analyze our results in Table I, we observe that there is good agreement between our results and previous calculations [13].
3.2. Electronic properties
Band structure
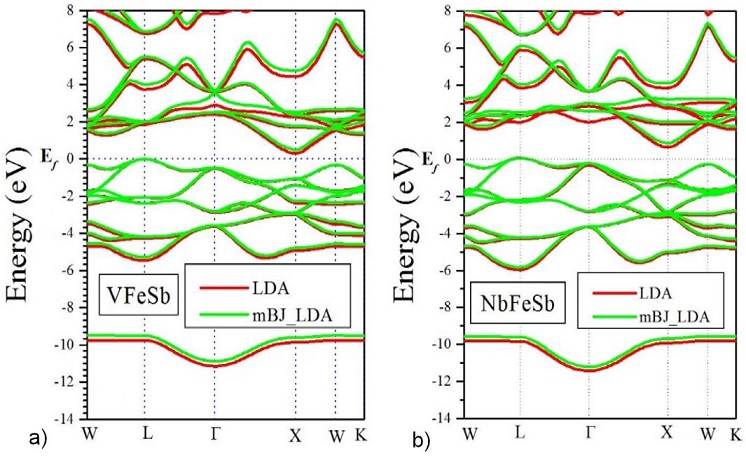
In the present work, we study the energy band structure at equilibrium lattice parameters and following that our materials have a very narrow gap, we have used the local density approximation proposed by Perdew and Wang [14] and the modified Becke-Johnson potential [15]. Figure 3 displays the band structure of VFeSb and NbFeSb, respectively, with LDA approximation and with mBJ. In Table II, we present the gap value obtained from our band structure calculations using LDA and the new mBJ along with previous theoretical results. The calculated energy band structure, at equilibrium lattice parameters with the mBJ-LDA, gives us the band gap shown in Fig. 3; 0.583 eV for VFeSb and 0.991 eV for NbFeSb. These values are better than those calculated by the local density approximations. The band gap calculated with the LDA is 0.302 eV for VFeSb and 0.543 eV for NbFeSb and is compared in Table II to other calculated values [4].
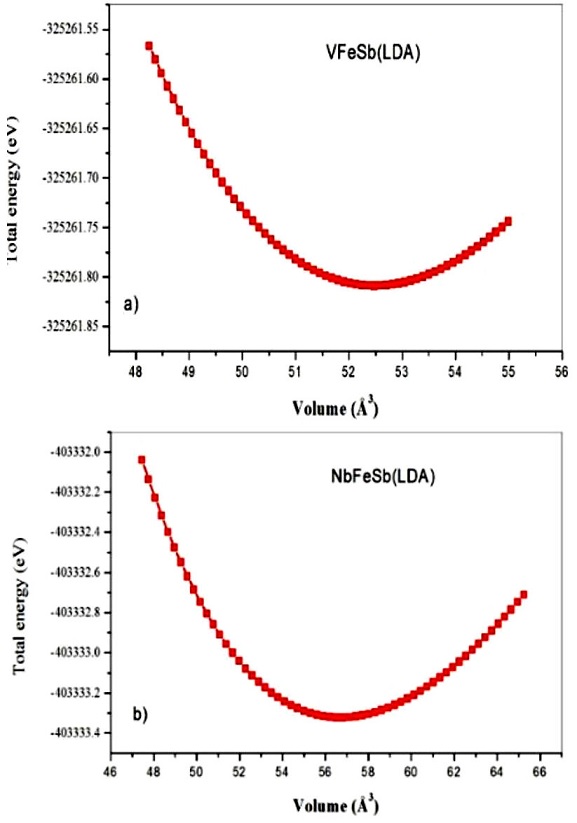
FIGURE 2 Variation of the total energy as a function of the atomic volume of the half-Heusler compound a) VFeSb and b) NbFeSb with the LDA.
Density of states (DOS)
The character of the band states for these materials can be defined by calculating the total and partial state densities DOS using the Tetrahedron method, which was developed by Blöck et al. [16] which requires a large number of special points in the irreducible Brillouin zone.
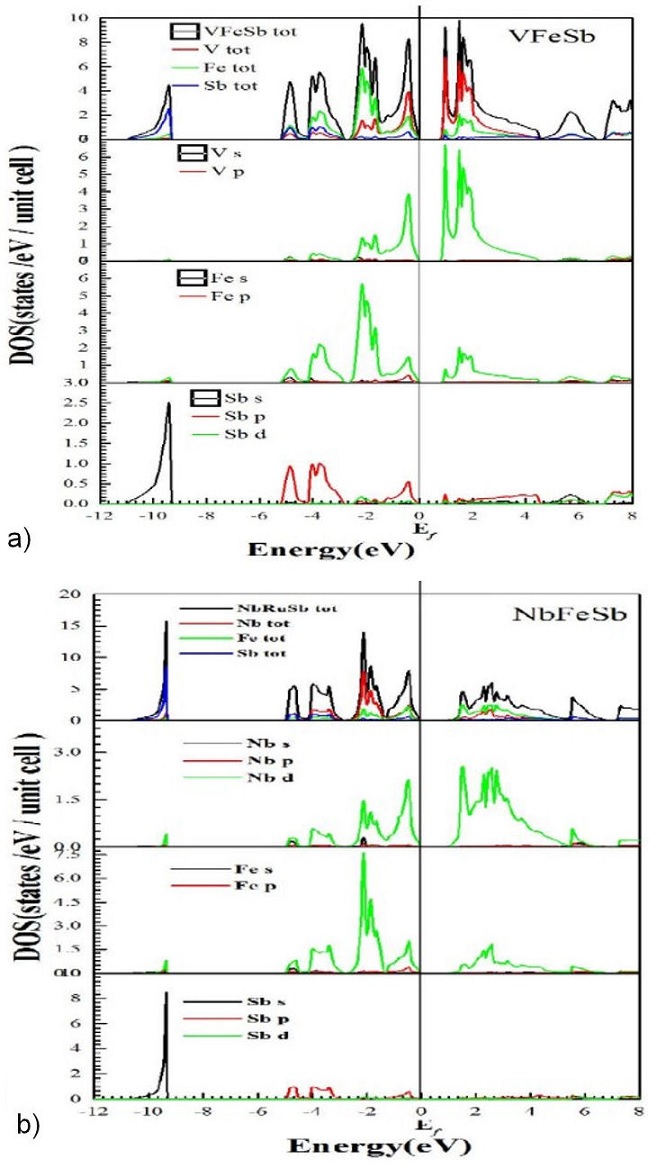
Figure 4 shows the total densities of states (TDOS) and partial densities of states (PDOS) of the VFeSb and NbFeSb compounds calculated by respecting the references of Fermi energy levels which correspond to 0 eV. We note the presence of a small gap between the binding states and the anti-binding states in the two VFeSb and NbFeSb. This small gap corresponds to the character of semiconductor compounds. As we can say that between -5 eV and 0 eV (Fermi level); It’s a hybridization of d-states of Vanadium (V) and d-states of Iron (Fe) and P states of Antimony (Sb). From this analysis. The intensity of the peaks decreases when the V is replaced by Nb; Note that all the DOS state densities of the two compounds are similar and come with equal contributions from all the elements X (X: V, Nb) Antimony (Sb) its strong contribution comes from the s orbital between the interval -12 eV and -9 eV and with and essentially of the p orbital between -5eV and the Fermi level; Above the Fermi level, the density of state is made up of a mixture of s, p orbitals and d. In the vicinity of the Fermi level region, the major contribution to the DOS is due to the d orbital of both Iron (Fe) atoms and (V and Nb). As we can say that between -5 eV and 0 eV (Fermi level); It’s a hybridization of d-states of Vanadium (V) and d-states of Iron (Fe) and P states of Antimony (Sb). From this analysis, we can suggest that the d states of the electrons of the Iron atoms and (V and Nb) will play a major role in the transport properties. The phonon dispersion of the compounds: VFeSb and NbFeSb have already been studied by other researchers; [3-17-18].
3.3. Optical properties
In our calculations, we will use the structural and electronic properties for our materials already calculated previously and summarized in Table I and II [4].
The Dielectric function ε(ω) has real and imaginary parts,
The cubic crystal structure of the VFeSb and NbFeSb half-Heusler compounds only requires the calculation of a single dielectric tensor component to characterize the imaginary part of the dielectric.
For our calculations, we use the exchange-correlation potential MBJ [19].
The imaginary part of the dielectric function iε 2(ω) is given by:
The real part of the dielectric tensor ε 1(ω) is then determined using the Kramers-Kronig relation
The calculation of important optical functions such as the refractive index n(ω), the extinction coefficient k(ω) and energy loss function L(ω) is performed using the following expressions [20]:
The study of optical properties of materials is very important to define the internal structure of the VFeSb and NbFeSb compounds. The real and imaginary parts of the dielectric function, ε 1(ω) and ε 2(ω),
refractive index n(ω), and loss function L(ω) are given in Table III and shown in Figs. 5-7 as functions of the photon energy in the range of 0-30 eV. The imaginary part of the dielectric function ε 2(ω) gives the information of absorption behavior of VFeSb and NbFeSb. The threshold energy of the dielectric function occurs at E 0 = 0.329 eV for VFeSB and E 0 = 0.520 eV for NbFeSb, where these energies correspond to the fundamental gap at equilibrium. It is well known that the materials with band small gaps. From Fig. 5, for the imaginary part, it is clear that there are strong absorption peaks in the energy range of 0.329-30 eV (VFeSb) and 0.520-30 eV (NbFeSb). The maximum absorption peak for these materials are, respectively, at 46.66 eV and 44.013 eV. The peak around 1.25-25 eV for VFeSb and 185-25 eV for NbFeSb appear due to the electronic transition from state of the valence band (VB) to the unoccupied state in the conduction band (CB). The real part of the dielectric function ε 1(ω) is also displayed in Fig. 5. This function ε 1(ω) gives us information about the electronic polarizability of a material.
TABLE III Optical dielectric constant ε(0) and refractive index n(0) of VFeSb and NbFeSb at zero photon energy.
| Material | ε(0) | n(0) |
| VFeSb | 26.89 | 4.13 |
| NbFeSb | 26.92 | 4.31 |
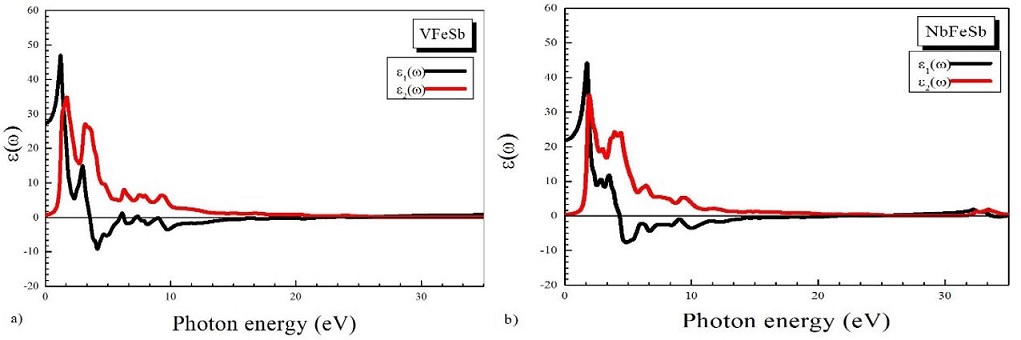
FIGURE 5 Real and imaginary parts of the dielectric function ε(ω) for a) VFeSb and b) NbFeSb with the mBJ-LDA.
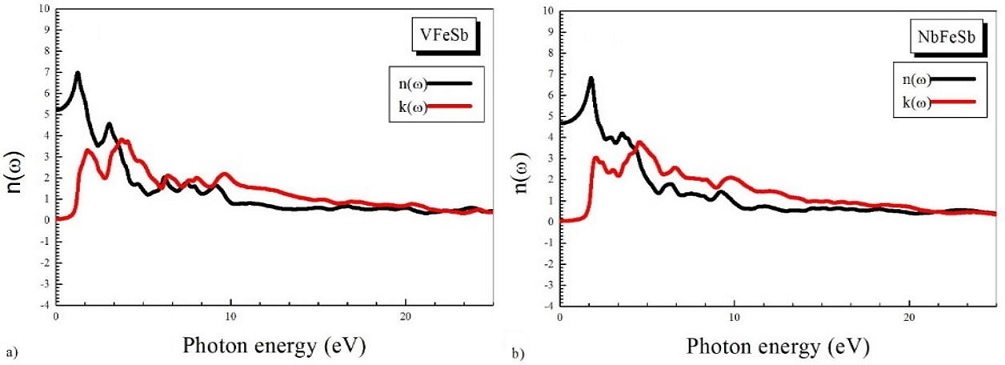
FIGURE 6 Refractive index n(ω) and extinction coefficient k(ω) for a) VFeSb and b) NbFeSb with the mBJLDA.
The static dielectric constant in its zero frequency limit was found to be ε(0) = 26.89 and ε(0) = 20.92 for VFeSb and NbFeSb, respectively. The value of ε 1(ω) increases as the photon energy increases and reaches a maximum before dropping to a value below the Fermi level, see Fig. 5. The refractive index is displayed in Fig. 6. The static refractive index n(0) is obtained from the real part of the dielectric function, namely,
and is found to be 4:1325 and 4:3123 for VFeSb and NbFeSb, respectively.
The refractive index is larger than 1 as photons penetrate to the material; they are slowed down by the interaction with electrons.
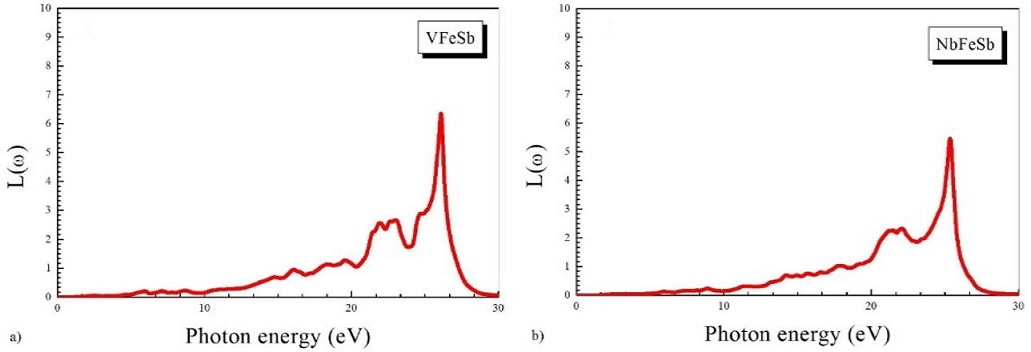
A refractive index of less than unity shows that the group velocity (V g = c/n) of the incident radiation is greater than c. The energy loss function is displayed in Fig. 7. The energy loss function L(ω) is an important quantity that describes the energy loss of a fast electron traversing in a material. The peaks in the L(ω) spectra characterize the plasma resonance. We can see that the main sharp structure of L(ω) is located at about 15 eV, corresponding to a rapid decrease of reflectance. The resonant energy loss is seen at 26.11 eV and 25.37 eV for VFeSb and for NbFeSb.
3.3.1. Thermal properties
We used the quasi-harmonic Debye model (realized in the GIBBS computer code and integrated in the WIEN2k computational package) [21]:
where M is the molecular weight per unit volume, B S is the module of the compressibility [22]
and f(σ) is given by the following function [23,24]:
We obtain the pressure and temperature equilibrium by the minimization of the Gibbs function:
The heat capacity at constant volume C V [25] is
The analysis of the thermal properties of materials provides information about their phase stability, melting point, strength, bonding nature, etc. In this work, we calculated the thermal properties of the intermetallic half-Heusler, ternary compounds (for VFeSb and NbFeSb) using a density functional theory based approach. To determine the thermal properties, the quasi-harmonic Debye model (realized in the GIBBS computer code and integrated in the WIEN2k computational package) is used. The obtained thermal properties, at room temperature, for the for VFeSb and NbFeSb ternary compounds are presented in Fig. 8. It is seen that the for VFeSb and NbFeSb, ompounds presents the highest heat capacity among the three compounds, as observed in Table IV. On the other hand, the value of the heat capacity (Cv) at higher temperature for all the three crystal structures reaches the value of the classical limit of Dulong and Petit, (69.04 J/molK for VFeSb and 71.22 J/molK for NbFeSb).
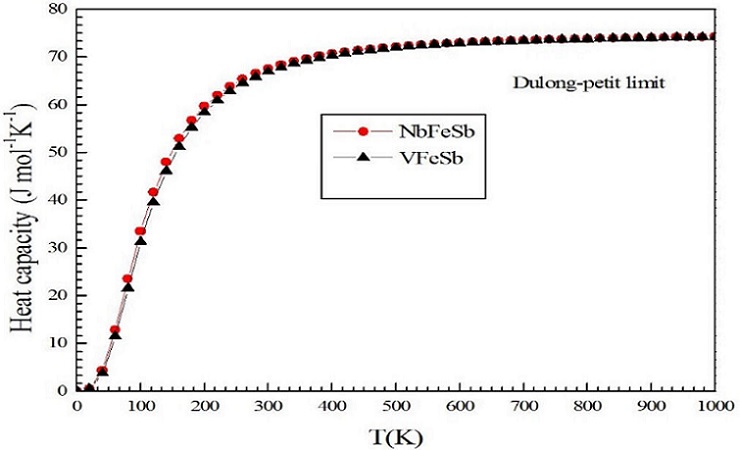
TABLE IV The selection of the thermal properties at 300 K: thermal expansion coefficient (a in 10−5 K−1): (Cv and Cp in J/mol K), isothermal and adiabatic bulk moduli (B and BS , in GPa), Debye temperature (θD in K) for VFeSb and NbFeSb at zero frequency.
| Material | CP | CV | B | BS | θD |
| VFeSb | 66.03 | 69.04 | 191.45 | 195.34 | 501.22 |
| NbFeSb | 67.11 | 71.22 | 189.23 | 193.15 | 550.00 |
4. Conclusion
We have presented a theoretical analysis of the different structural, electronic, optical, and thermal properties of VFeSb and NbFeSb compounds using the FP-LAPW method with the LDA and mBJ-LDA. The results of the band structure show that the XFeSb (where X = V, Nb) compounds with the maximum valence band and the minimum conduction band at the L and X symmetry points, respectively, are indirect band-gap semiconductors,which is in good agreement with the previous available studies. The results observed for the density of states may be due to the electro-negativity values (according to the Pauling scale, corresponding to the V, Nb, Sb and Fe atoms, respectively, where they are in close proximity with each other). The outermost electrons of the Sb atom appear to be polarized to some extent in the direction of the Fe atom. The optical properties such as dielectric function, refractive index, and electron energy loss are studied in the energy range of 0-35 eV. The above properties of VFeSb and NbFeSb have provided the appearance of several peaks associated with the interband transitions in the two structures. The thermal properties presented in Table IV showcase that our compounds present high Debye temperatures, which indicates that they may present important thermal conductivities. In our calculations the magnetic properties are not considered because our compounds have a small gap, corresponding to a semiconductor behavior, i.e., with vanishing magnetic moments.

