











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
1.Introduction
Tha MAX phases, the extension of the Hägg phases known since 1960 when Nowotny reported the discovery of more than 100 carbides and nitrides 1,2, represent an exceptionally extensive class of ceramics. They correspond to a general formula of the type: Mn+1 AXn where M is a transition metal (Ti, V, Cr), A is a metal in general groups IIIA or IVA (Al, Si, P, S) and X is either C and/or N, and n = 1, 2, 3 2,3. Despite their relatively old discovery, their physical properties have been relatively little studied and it was not until 1996 that a systematic work of synthesis and characterization was undertaken by an American team from Drexel University (Philadelphia) led by Mr. Barsoum 4. Most of the MAX phases are of M2AX stoichiometry with the space group P63 / mmc 5. Figure 1 shows the crystal structure of Sc2SiX. These M2AX has attracted more attention due to the fundamental properties, usually associated with both metals and ceramics 6. In general, MAX phases showcase a metallic behavior 7-12, are good electrical and thermal conductors 13, have a high elastic modulus 13, are machinable 14, resitant to oxidation, tolerant to damage, elastically rigid, and present low thermal expansion 15. Additionally, they have excellent thermal shock and corrosion resistance 16,17. In addition, MAX phases are used as the replacement of machinable ceramics, furnace cabinets, wear and corrosion protection, heat exchangers, applications in which rotating parts are used, low friction applications based on the lubricating power of the basal plane 18. In this work, the idea of the existence of a new class of superconducting materials. Despite all the efforts, it is obvious that the Sc2SiC and Sc2SiN MAX phase’s compounds have not been subjected to theoretical and experimental studies. The purpose of our work is to calculate and study the structural, electronic, and elastic properties of the compounds Sc2SiC and Sc2SiN MAX phases, using first-principle calculations of density functional theory (DFT) within the full-potential linearized augmented plane-wave (FP-LAPW) approach. This document is organized as follows: computational details are described in Sec. 2, the results are discussed in Sec. 3, and finally, gives the conclusions.

2.Computational detail
Today, there are several codes with a wide variety of approximations that we can make use of for a theoretical study. In our calculations, we used the first-principle methods of density functional theory (DFT) based on the full-potential linearized augmented plane-wave (FP-LAPW) approach as implemented in WIEN2K code 19,20. This application allowed us to study the structural, electronic, and mechanic properties of max phase types Mn+1 A X n or M = Sc, A = Si, (X = C, N), n = 1-3 1,2. The algorithm is based on the density functional theory (DFT) within the local density approximation (LDA) proposed by Perdew and Wang 21 for the exchange correlations functional. It calculates the self-consistent solution of the equations of Kohn and Sham 22. The electronic configurations of the sets of the system studied are Sc:[Ar]4s2 4d1, Si:[Ne]3s2 3p2, N:[He]2s2 2P3 and C:[He]2s2 2P3. We have chosen radii Rmt such that there will be no overlap of the Muffin-Tin spheres; the values used are 2.6, 1.9, 2.8,1.6 for the Sc, Si, C, and N atoms, respectively. The number of points k used in the integration of the first Brillouin zone is (1500 k-points) for our structure this integration of k on the Brillouin zone is carried out using the Monkhorst and Pack of mesh 23, and Rmt*Kmax is taken equal to 7 (where Rmt represents the smallest radius Muffin-Tin and Kmax the cut-off of plane waves), within the spheres, the wave functions of the valence region extend to lmax=10 .
3.Results and discussion
3.1.Structural properties
Both Sc2SiC, Sc2SiN compounds crystallize in the Cr2AlCd crystal structure. The positions of atoms in Sc2SiX, (X = C, N) are as follows: C atoms are placed at the positions (0, 0, 0), the Si atoms are at (1/3,2/3,3/4) and the Sc atoms are at (1/3, 2/3, ZM) 12, where z is the internal free coordinate. We have first minimized the internal parameters ZM by taking random lattice parameters to start our calculations because of the lack of any a priori information. We then performed detailed structural optimizations by reducing all energies to a minimum. Our results of the calculated lattice constants α and c, bulk modulus and its pressure derivative, and the optimized free internal parameters obtained with the LDA functional at 0 K are reported in Table I for Sc2SiC and Sc2SiN. These properties were determined by adjusting the total energy as a function of volume, using the Murnaghan equation 24. The report (c/α) is in good agreement with the ideal values (Z =1/12= 0.0833) and with the theoretical compactness report (c / a = 4.89) 8. From the results of , we can say that the compound Sc2SiN is harder and more stable than Sc2SiC due to the high value of the compressibility modulus and it has minimum energy. We note that upon the substitution of C by N, the values of α and c increased slightly, a consequence of the electronegativity of nitrogen being larger than that of carbon. The bulk modulus increases by 13.73% as the C is substituted with N.
Table I The equilibrium lattice parameters (a; c, and c=a), equilibrium volume, internal parameter Z(M), bulk modulus (B0) in (GPa) and its pressure derivative (B’) for Sc2SiX (X = C, N).
| A(Å) | C(Å) | c/a | B0 | B´ | Z(M) | V0Å3 | |
| Sc2SiC | 3.18 | 13.52 | 4.24 | 130.7 | 4.03 | 0.0893 | 116.76 |
| Sc2SiN | 3.16 | 13.36 | 4.26 | 151.5 | 4.12 | 0.0886 | 108.29 |
3.2.Formation and cohesive energy
To see the relative phase stabilities, we calculated the energy of formation for Sc2Si X,X=(C,N) using 5,25
where x, y, and z indicate a
number of atoms in a unit cell, of Sc, Si, N, and C atom in cell respectively.
where
3.3.Electronic properties
3.3.1.Band structures
We calculated the electronic band structures of Sc2SiC and Sc2SiN at equilibrium lattice parameters along with the high symmetry directions in the first Brillouin zone, with LDA, GGA, and GGA+TB-mBJ, which are presented in Fig. 2, 3 , and 4, respectively. The conduction bands are shifted towards the valence bands and overlap significantly at the Fermi level, and the absence of a gap for the two compounds clearly indicates the metallic character. The states with energies below minus -10 eV, -14 eV below the Fermi level provided (C, N) -2s states respectively, the states just at the Fermi level were mainly Sc-3d states, (C, N) -2p and Si-3s and 3p. This result confirms the metallicity of these two materials.
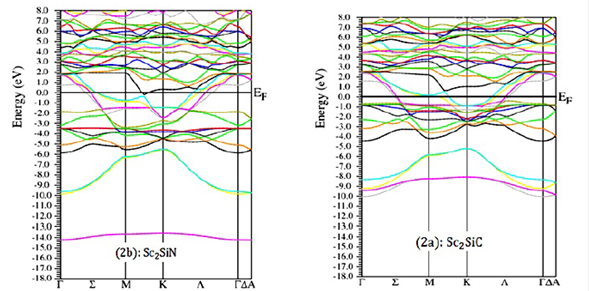
Figure 2 Calculated band structure of a) Sc2SiC and b) Sc2SiN with LDA approximation along the high symmetry directions in the Brillouin zone at ambient conditions.
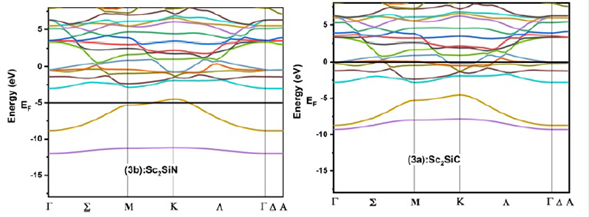
Figure 3 Calculated band structure of a) Sc2SiC and b) Sc2SiN with GGA approximation along the high symmetry directions in the Brillouin zone at ambient conditions.

3.3.2.Densities of states
We have calculated the total and partial density of states (DOS) presented in Fig 5a, 5b. We see that there is no band gap Eg at the Fermi level for the two materials, which allows us to deduce that these two compounds have a metallic bonding nature since the DOS has a large finite value at the Fermi level. Thus, the broad values of the Fermi energies (EF) (see Tabla II) confirm this result. At the level of Fermi, the DOS is 2.025 and 1.695 states per unit cell per eV for Sc2SiC and Sc2SiN respectively. It can be concluded that Sc2SiN is more conductive than Sc2SiC. On the other hand, Sc-3d electrons play the dominant role in DOS, which primarily contributes to DOS at the Fermi level and should be involved in the conduction properties. Although 3d electrons are generally considered as low-efficiency drivers. The C-2p and Si-3p electrons do not significantly contribute to DOS at the Fermi level and are therefore not involved in the conduction properties. The partial DOS profiles in Fig 5a, 5b show another interesting feature: the hybridization peak is strongly dominated by the contribution of the (C, N) 2 -p, Sc-3d states, but there is weak hybridization of the Si-3p and Sc-3d states; this would be beneficial to the structural stability of Sc2SiN. The main difference is that the electrostatic attraction of nitrogen is higher than that of carbon, and the electrical conductivity of Sc2SiN is greater than that of Sc2SiC. It can be concluded that the Sc-3d, Si -3p bonds are stronger in Sc2SiN than in Sc2SiC.

Table II The calculated values of the energy of formation (EFor), bulk modulus (B0), cohesive energies (ECoh),) and the valence electron concentration (val-el) for Sc2SiX (X = C, N).
| Sc2SiC | Sc2SiN | |
| EEq | -7407.880848 | -7474.654553 |
| EFor(eV/atom) | -0.72 | -3.6 |
| ECoh(eVatom) | 7.07 | 4.98 |
| valence electron | 60 | 62 |
| B0 (Gpa) | 130.7 | 151.5 |
| EF | 0.51 | 0.60 |
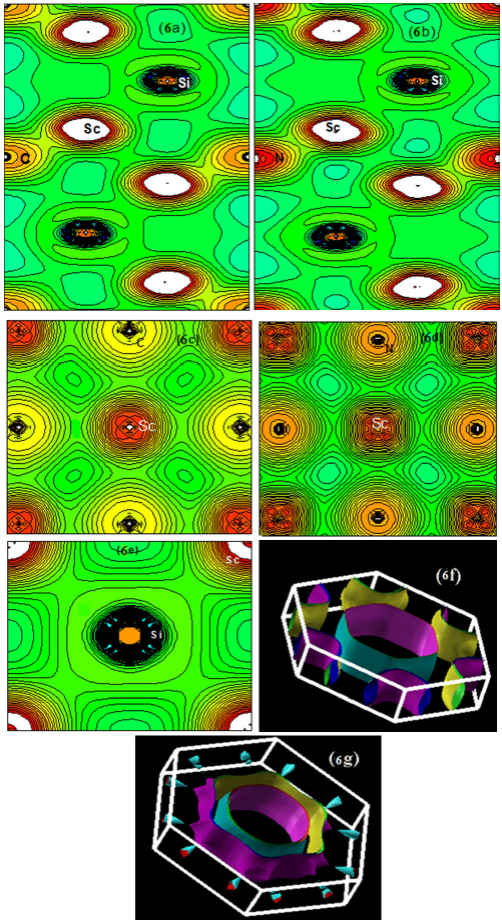
3.3.3.Charge densities and Fermi surface
As a rule of thumb, the nature of the chemical bond is related to the
difference in electronegativity between the elements at play, as it informs
us about the charge transfer and, consequently, on the nature of the bond in
the materials. The MAX phases are generally stacks of a “hard” M-X bond and
a “soft” M-A bond in the c direction. The Ti-C bond strength is much
stronger than the Ti-Al bond in Ti 2AlC 31. Figures
6a and 6b show the contours
of the charge densities of Sc2Si X (X= C, N) in the (11
3.4.Elastic properties and mechanical stability
The calculation of the elastic constants will make it possible to examine the mechanical stability of the ground state proposed by the FP-LAPW method. The elastic behavior of a hexagonal system is described by five independent constants C11, C33, C44, C12, and C13 and the sixth constant C66 is calculated from C11, C129. These constants can be determined from a change of total energy as a function of the constraint. The evaluation of elastic constants was made from deformation formulas proposed by Wallace. We also find that the elastic constants of the different phases satisfy the following relation 11,32.
Calculations of second-order elastic constants of Sc2Si X, X = (C, N) are presented in Table III. It can be said that the compounds Sc2Si X, X = (C, N), are mechanically stable because all these elastic constants are positive and meet the criterion of mechanical stability 11 on top of the fact that all the elastic constants increase when C is replaced by N. Thus, it can be concluded that the Sc-N bonds are stronger than the Sc-C bonds. The modules C ij have a heavyweight in the study of materials, especially module C44. From this quantity, several properties can be determined such as brittleness, ductility, among others. The two constants C11 and C33 are connected to the directions α and c, respectively, while C44 is related to the shear constraint. The module of C44 shows that Sc2SiN is harder than Sc2SiC, a result that is similar to that found in the optimization part.
Table III Elastic constant C ij for Sc2SiX (X = C, N).
| C11 | C12 | C13 | C33 | C44 | C66 | B/C44 | |
| Sc2SiC | 224.62 | 77.3 | 73.67 | 309.86 | 100.92 | 73.66 | 1.32 |
| Sc2SiN | 263.68 | 75.14 | 87.92 | 330.43 | 127.47 | 94.27 | 1.18 |
The modulus B, the shear modulus G, Young’s The modulus E, the Poisson ratio for
Sc2SiX, X = (C, N), get from the individual elastic constants by
the Hill approximation, this approximation is based on the approaches of Reuss
and Voigt are given In Table IV, with
B=(BV+BR) = BH (Hill’sbulk modulus) and
G=(GV+GR)=GH (Hill’s shear) 31,32. The Young’s modulus E and
the Poisson’s ratio 𝑣 are determined by the relations E=9 BG/(3 B+G) and
The Sc2SiN compound is stiffer and harder than Sc2SiC due to the high value of the Young’s modulus and compressibility modulus.
From the Pugh Criterions 37, a material must behave ductile if
An additional point for the brittle/ductile conduct of this phase results from the evaluated Poisson’s ratio. According to Frantsevich et al. 38, metals with a Poisson’s ratio of about 1/3 are ductile, whereas metals with a Poisson’s ratio of less than 1/3 are deduced to be fragile; the values for Sc2SiC and Sc2SiN are 0.22 and 0.23, respectively.
Another useful factor is the machinability index
We have determined the elastic anisotropy factors, A1, A2 and A3 for the hexagonal crystal determined by the ratio between the linear compressibility coefficients along with the α- and c-axis. There are three independent elastic shear constants for hexagonal crystals; thus, three shear-type anisotropy factors can be determined by 11
Table IV The bulk modulus B, shear modulus G, Young’s modulus E (all in GPa), Poisson’s ratio v, anisotropic factor A, linear compressibility ratio f = Kc=Ka for Sc2SiX (X = C, N).
| E | B | G | A1 | A2 | A3 | v | f | G/B | |
| Sc2SiC | 219.23 | 133.09 | 89.45 | 1.04 | 1.37 | 1.4 | 0.22 | 0.65 | 0.67 |
| Sc2SiN | 263.52 | 149.99 | 109.14 | 0.85 | 1.35 | 1.15 | 0.207 | 0.67 | 0.73 |
These anisotropy factors are presented in Table
IV, and any deviation of more than or less than 1 corresponds to an
elastic anisotropy. The magnitude of the deviation from 1 is a measure of the
degree of elastic anisotropy possessed by the crystal 18. Finally, the ratio between the linear
coefficients of compressibility
The results show that for Sc2SiC and Sc2SiN, f =(0.65,0.67), respectively. Furthermore, since these values of f are less than 1, the compressibility along the c-axis is smaller than alogn the α- axis for both compounds. For comparison, this parameter for the isoelectronic Nb2SnC compound is f = 0.94, i.e., it lies close to the isotropic limit f = 1 12,33,40.
4.Conclusion
In summary, using the augmented planar wave (FP-LAPW) method, based on the DFT, within the LDA, GGA, and GGA+TB-mBj. We studied the structural, electronic, and elastic properties of Sc2SiC, Sc2SiN compounds. Our results show that the substitution of C by N in Sc2SiC affects the structural, elastic, and electronic properties of the material. We calculated both formation energies of the carbon-based and the nitrogen-based compounds, which allow us to conclude that the synthesis of these two compounds can be realized. The state Sc-3d and C-2p are stronger than the state Sc-3d and Si-3p. Sc2SiX, X = C, N, has a metallic-covalent-ionic character in nature. Fermi’s surface properties were studied for the first time, and the replacement of N by C increases all elastic constants. The elastic anisotropy of Sc2SiN is higher than that of Sc2SiC.

